the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 11 Feb 2026
| 11 Feb 2026
TChem-atm (v2.0.0): scalable performance-portable multiphase atmospheric chemistry
Oscar H. Díaz-Ibarra
Samuel G. Frederick
Jeffrey H. Curtis
Zachary D'Aquino
Peter A. Bosler
Lekha Patel
Cosmin Safta
Matthew West
We present TChem-atm, a performance-portable approach that enables efficient simulation of chemically detailed and multiphase atmospheric chemistry on modern heterogeneous computing architectures. Unlike previous efforts that rely on architecture-specific code or focus exclusively on gas-phase chemistry, TChem-atm supports fully coupled gas–aerosol systems with execution across CPUs, NVIDIA GPUs, and AMD GPUs through the Kokkos programming model. It integrates the flexible multiphase capabilities of the Community Atmospheric Model Chemistry Package (CAMP) with the high-performance kinetic routines of TChem, and includes automatic Jacobian construction with support for a range of stiff ODE solvers. In a proof-of-concept integration with the particle-resolved model PartMC, TChem-atm reproduces the existing PartMC–CAMP implementation within solver tolerances and delivers substantial GPU speedups, especially for large particle populations. Performance benchmarks reveal substantial speedups on GPU platforms, particularly for large particle populations, with consistent results across hardware backends. TChem-atm enables performance-portable execution across CPUs and GPUs, though optimal efficiency may require modest architecture-specific tuning (e.g., team and vector sizes), with up to a twofold improvement on the NVIDIA H100. It directly supports sectional and particle-resolved host models, while modal aerosol schemes require minor adaptation to provide particle-scale quantities such as representative diameters. By enabling chemically detailed, multiphase simulations with performance portability and host-model flexibility, TChem-atm facilitates the incorporation of advanced chemistry into atmospheric models.
- Article
(4365 KB) - Full-text XML
- BibTeX
- EndNote




Simulating the chemical evolution of the atmosphere is central to predicting air quality, climate forcing, and ecosystem impacts. Atmospheric models must capture complex interactions between gas-phase reactions, aerosol microphysics, and multiphase chemistry – often at fine spatial and temporal resolutions and over extended simulation domains. As chemical mechanisms become more detailed and models increase in resolution, the computational cost of solving the associated systems of equations grows rapidly. To address this challenge, flexible and scalable methods are needed that can integrate complex chemistry into models while taking full advantage of modern computing architectures.
Simulating detailed gas-phase mechanisms, multiphase processes, and aerosol microphysics at such fine resolutions typically requires solving thousands of coupled, numerically stiff ordinary differential equations (ODEs) at each grid point. This makes chemical solvers one of the most computationally intensive components of atmospheric models. Graphics Processing Units (GPUs) offer a massively parallel architecture that can accelerate these calculations significantly, enabling more detailed and efficient simulations.
This acceleration is critical across a wide range of applications. In Earth system models such as E3SM (Energy Exascale Earth System Model), interactive chemistry and aerosols are essential for representing radiative forcing and cloud feedbacks over long timescales (Golaz et al., 2019; Rasch et al., 2019). In global chemical transport models like GEOS-Chem, GPU acceleration enables higher spatial resolution and real-time data assimilation (Bey et al., 2001; Eastham et al., 2018). Operational air quality models such as CMAQ must meet tight runtime constraints while still representing complex chemical and physical processes (Byun and Schere, 2006; Appel et al., 2021). Across these domains, reducing the computational burden of atmospheric chemistry is essential for improving both fidelity and performance.
Recent work has demonstrated the potential of GPU acceleration for atmospheric models. For example, Ruiz et al. (2024) achieved a 35× speed-up by distributing the CAMP solver load across GPU threads, and Alvanos and Christoudias (2017) reported kernel-level speed-ups of up to 20.4× for CUDA-based kinetic integration in EMAC. Cao et al. (2023) showed more than 1000× speed-ups for GPU-accelerated advection in CAMx using hybrid GPU–MPI strategies, while Quevedo et al. (2025) demonstrated that GPU-enabled CMAQ simulations could cut runtimes nearly in half. Similarly, Sun et al. (2018) highlighted the importance of memory layout and GPU-aware optimization in achieving efficient Rosenbrock solver performance in CAM4-Chem.
While these studies underscore the promise of GPU-accelerated atmospheric chemistry, most rely on architecture-specific APIs (e.g., CUDA) and hand-optimized kernels tightly coupled to the host model. This approach limits code portability, increases integration complexity, and hinders broader adoption of GPU acceleration across diverse computing environments.
To address these limitations, performance-portable approaches such as TChem have emerged, leveraging libraries like Kokkos to abstract architecture-specific details (Kim et al., 2023). TChem provides a scalable and efficient backend for evaluating gas-phase and surface reaction kinetics, thermodynamic properties, and Jacobians via automatic differentiation. However, TChem was originally developed for gas-phase chemistry and did not natively support multiphase processes, limiting its application in comprehensive atmospheric chemistry models.
In parallel, we developed the Chemistry Across Multiple Phases (CAMP) library as a runtime-configurable tool for simulating gas- and aerosol-phase chemistry in models with varying aerosol representations (Dawson et al., 2022). CAMP decouples chemical logic from the host model's aerosol structure and supports modular chemical mechanism configuration and solver abstraction. However, while CAMP is flexible in mechanism design, its reliance on host-side solvers limited its scalability and portability to new hardware architectures.
Recognizing their complementary capabilities, the present work combines CAMP's support for multiphase chemistry with TChem's performance-portable computational backend. We refer to this extended version as TChem-atm, which is maintained as a standalone code with its own repository. TChem-atm builds on the software infrastructure of TChem (v2.0), which was originally developed for gas-phase kinetics in combustion applications. TChem provides the foundational components that TChem-atm inherits, namely the kinetic-model data structures, automatic and numerical Jacobian construction, and the batched-evaluation framework implemented with Kokkos. TChem-atm extends this base by introducing atmospheric-chemistry capabilities not present in TChem, including (i) support for aerosol- and multiphase reaction mechanisms, (ii) integration with CAMP's gas–aerosol process abstractions, (iii) an aerosol-model constant-data object and associated particle/section/mode interfaces, and (iv) mechanism parsing for heterogeneous and condensed-phase chemistry. These additions transform TChem's gas-phase engine into a performance-portable multiphase chemistry library suitable for atmospheric models across CPUs and GPUs. TChem-atm further provides interoperability with multiple ODE solvers, including TINES (Kim and Diaz-Ibarra, 2021) and SUNDIALS (Balos et al., 2021; Gardner et al., 2022; Hindmarsh et al., 2005, 2025).
TChem-atm is compatible with a wide range of aerosol representations, but the degree of direct interoperability differs across host models. Sectional and particle-resolved frameworks map naturally onto TChem-atm's computational-particle abstraction. Modal aerosol schemes, however, typically do not expose particle-scale quantities such as representative diameters or per-species mass vectors, and therefore require modest adaptation to supply these inputs. Once provided, TChem-atm can evaluate multiphase chemistry within modal frameworks in the same manner as for other aerosol representations. While both CAMP and TChem provide flexibility in chemical mechanism configuration, CAMP offers a structure tailored for gas–aerosol interactions, whereas TChem enables efficient execution on heterogeneous architectures via the Kokkos programming model.
This paper presents the first application of this integrated library to atmospheric modeling, with a focus on enabling efficient aerosol–chemistry interactions in high-resolution simulations. The present study is intentionally scoped as a proof-of-concept implementation that validates correctness and establishes performance characteristics of the new multiphase chemistry framework. Because the integration of CAMP chemistry with TChem's performance-portable backend is novel, our immediate priority is demonstrating functional equivalence to existing implementations and quantifying solver and hardware scaling. Application within regional- or global-scale models is a natural next step, but lies beyond the scope of this initial paper. The ability to interchange solvers across platforms without reimplementing solver logic streamlines deployment and optimization. By supporting scalable chemistry calculations on diverse architectures, this work represents a concrete step toward the generalized aerosol/chemistry interface advocated by Hodzic et al. (2023).
Although TChem-atm achieves performance portability across CPUs and GPUs without architecture-specific code, performance is further improved when tunable parameters (e.g., Kokkos team and vector sizes) are optimized for the target hardware. As demonstrated in Sect. 3.2, architecture-aware tuning yields up to a factor-of-two speed improvement on the NVIDIA H100 relative to the Kokkos default configuration. We note that while our current implementation succeeds as a first step toward performance portability, individual applications could be tuned for even greater performance. Such tunings would depend on the chemistry mechanisms present in the application, the choice of solver, and the specific computational architecture. Here, we introduce the capability to flexibly define atmospheric chemistry mechanisms for a variety of applications in a common computational environment, capable of running on a variety of advanced computing architectures; optimized tunings for specific applications, solvers, and architectures is a subject marked for future work.
TChem-atm is a performance-portable chemistry library designed to support multiphase atmospheric chemistry across a range of computing architectures. Figure 1 presents a high-level overview of the TChem-atm library and outlines the structure of this section.
We begin with an overview of the software architecture and the relationship between TChem, CAMP, and TChem-atm (Sect. 2.1), since these design choices define the core abstractions used throughout the implementation. The workflow then begins with user-defined YAML input files that specify gas-phase chemical mechanisms and aerosol processes (Sect. 2.2). These inputs are parsed into structured data objects for the gas and aerosol phases (Sect. 2.3), which are synchronized to device memory via dual Kokkos views. TChem-atm then constructs source terms for gas-phase chemistry (Sect. 2.5) and for aerosol-phase processes using a flexible abstraction that supports sectional and particle-resolved representations (Sect. 2.7 and 2.6). Performance portability is achieved through the Kokkos programming model (Sect. 2.8), which enables execution on CPUs as well as NVIDIA and AMD GPUs. If needed, Jacobian matrices are computed (Sect. 2.9) and passed to one of several supported ODE solvers (Sect. 2.10). Our testing approach is described in Sect. 2.11. Finally, we describe the integration of TChem-atm with PartMC, which enables particle-resolved aerosol simulations using batched, scalable multiphase chemistry (Sect. 2.12). The following subsections provide implementation details for each component of the implementation.
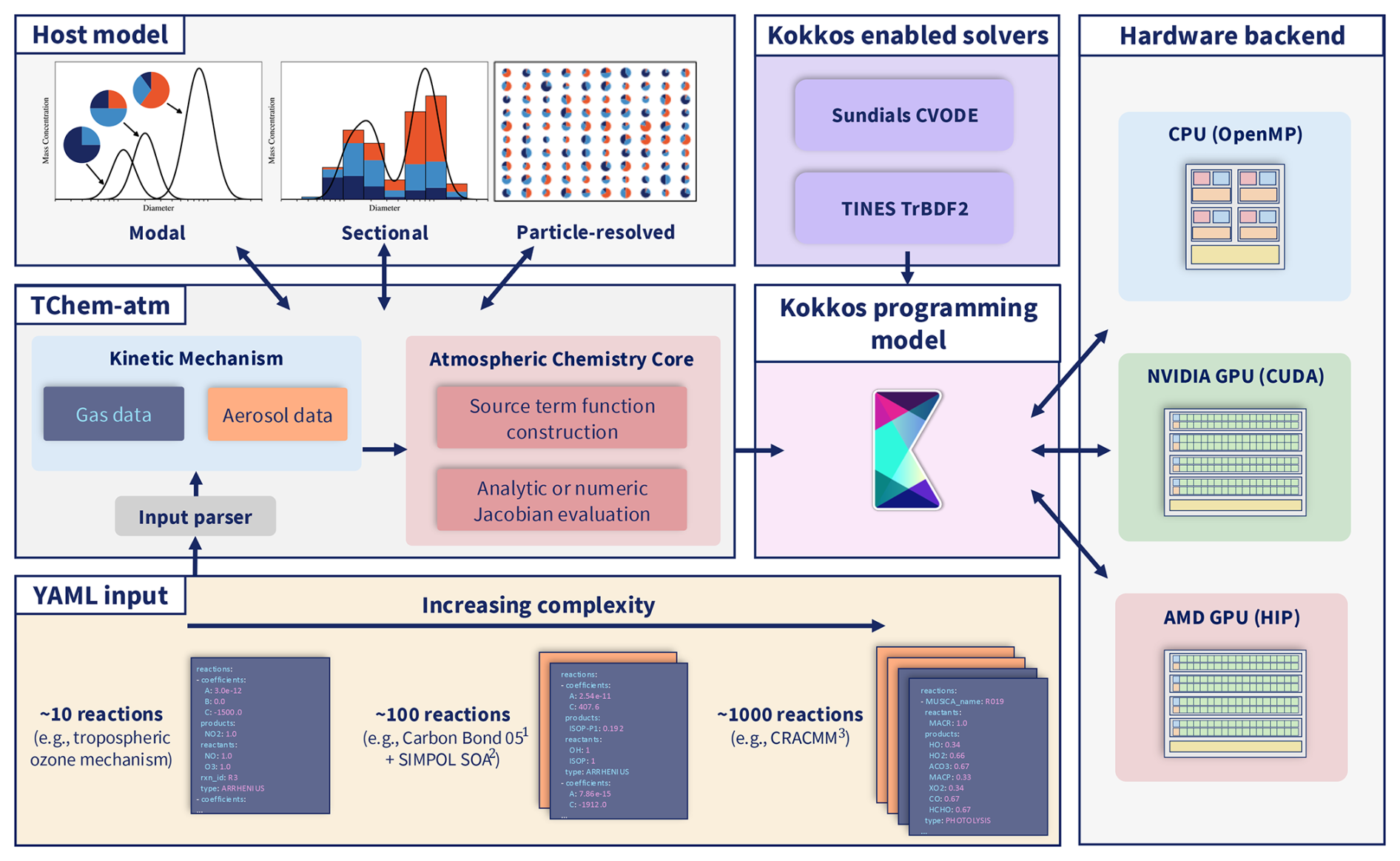
Figure 1Diagram of TChem-atm highlighting the customizability of the chemical mechanism and interoperability with numerous host model aerosol treatments. Further customization is supported for the selection of various Kokkos-enabled solvers, which allow performance portable computing across numerous platforms. 1 Carbon Bond 05: Yarwood et al. (2005); 2 SIMPOL: Pankow and Asher (2008); 3 CRACMM: Pye et al. (2023).
2.1 TChem-atm Provenance
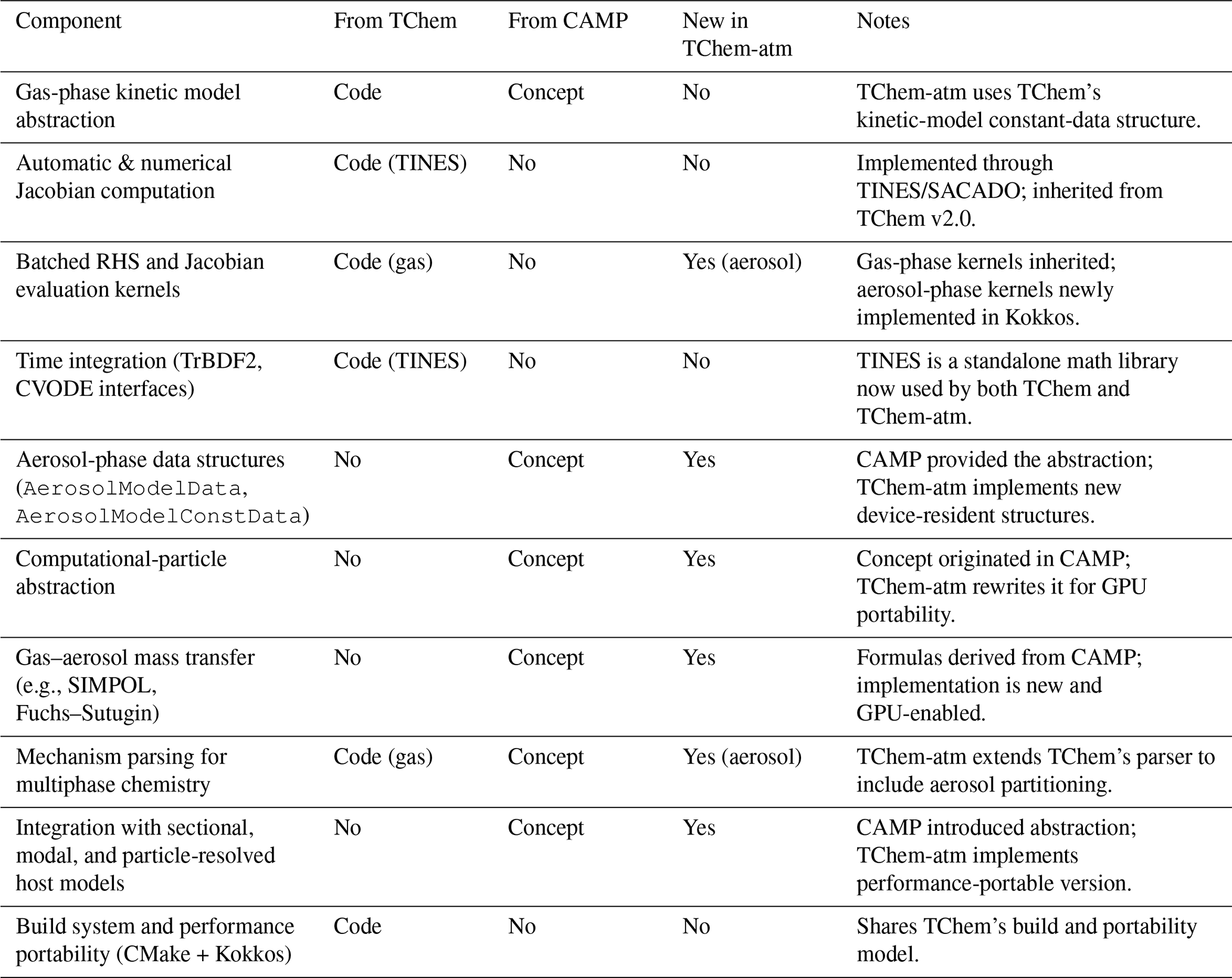
TChem-atm follows the software architecture of TChem v2.0, sharing its Kokkos-based parallel model, CMake-based build system, and mechanism-independent kinetic-model abstraction. The analytic and numerical Jacobian capabilities in TChem-atm are adapted directly from TChem and rely on TINES/SUNDIALS for time integration and Newton/Krylov linear algebra. What is new in TChem-atm compared to TChem is the multiphase extension: CAMP-style gas-aerosol partitioning are added, new data structures (AerosolModelData and AerosolModelConstData) are introduced, and the RHS kernels now account for gas–aerosol mass transfer. These atmospheric-chemistry extensions are not present in TChem and required new mechanism parsing, new source-term kernels, and a generalized notion of “computational particles” to support sectional, modal, and particle-resolved host models.
TChem-atm does not depend on TChem as an external library. All numerical functionality required for atmospheric chemistry – Jacobian construction, batched RHS evaluation, and stiff ODE integration – is now provided by the standalone TINES library, which was separated from TChem expressly to support multiple projects. Only a few legacy utility routines from TChem remain in TChem-atm; the combustion-specific components of TChem are not needed. This separation allows TChem-atm to evolve independently while sharing a common numerical backbone with TChem through TINES.
TChem-atm incorporates the multiphase-chemistry abstractions originally introduced in CAMP, but it does not embed CAMP directly nor merge the two code bases. CAMP provides a mechanism-agnostic representation of gas–aerosol processes (e.g., partitioning, heterogeneous chemistry, condensed-phase reactions) and a unifying “computational particle” interface that allows chemistry to operate consistently across modal, sectional, and particle-resolved host models. TChem-atm adopts these abstractions and reimplements the corresponding source-term formulations using its own Kokkos-based backend.
The specific components inherited conceptually from CAMP include: (i) the representation of aerosol–gas coupling processes (mass transfer, heterogeneous reactions, etc.); (ii) the use of mechanism files to define multiphase chemistry at runtime; and (iii) the computational-particle abstraction that enables TChem-atm to work with different aerosol frameworks.
What is new in TChem-atm is the implementation of these concepts within a fully performance-portable infrastructure, including: (i) new data structures for aerosol-phase species and their device-resident constant data (AerosolModelData/AerosolModelConstData); (ii) GPU-accelerated source-term kernels for gas–aerosol coupling; (iii) integration with TINES and Kokkos for batched multiphase ODE solves; and (iv) a unified atmospheric-chemistry mechanism parser that extends TChem's gas-phase parser to support multiphase processes.
Thus, TChem-atm should be viewed not as a code-level merge of CAMP and TChem, but as a new atmospheric-chemistry engine that inherits CAMP's abstractions conceptually while implementing them natively within the TChem/TINES performance-portable numerical framework.
Table 1Relationship between TChem, CAMP, and TChem-atm. Components are categorized as inherited as “Code” or “Concept”.
2.2 Chemical Mechanism Configuration
To support flexibility and ease of use, both CAMP and TChem-atm adopt human-readable configuration systems based on JSON or YAML for defining chemical mechanisms and model parameters (Dawson et al., 2022; Kim et al., 2023). This design enables users to specify complex reaction mechanisms, thermodynamic properties, and aerosol interaction parameters at runtime, without modifying the core codebase. Figure 2 shows an example gas-phase mechanism in YAML format for a simple system involving one reaction and three species (). This configuration is used to simulate the same chemical system independently across N computational cells, where each cell represents a distinct instance of the ODE system such as grid points in a host model or batches in a unit test.
The structured and editable format of these configuration files, along with the availability of validation tools, makes them accessible to a broad range of users – including those without specialized programming experience. In this work, we retain CAMP's approach for specifying multiphase chemistry processes such as gas–aerosol partitioning and condensed-phase reactions, and integrate it into the TChem-atm infrastructure. This unification allows researchers to flexibly configure chemically detailed multiphase systems in a portable, performance-optimized environment suited for both comprehensive atmospheric models and standalone simulations (e.g., chamber or flow-tube studies).

Figure 2Example YAML input specifying a simple gas-phase mechanism consisting of a single reaction with three species () used in simulations with N independent computational cells, each solving a separate instance of the ODE system.
2.3 Aerosol and Gas Kinetic Model Data
TChem-atm organizes the reaction mechanism information for gas and aerosol phases into two structured data objects as shown in Fig. 3. The gas phase data is stored in the Kinetic Model Constant Data (kmcd) object, while the aerosol phase data is stored in the Aerosol Model Constant Data (amcd) object.
Constructing these data objects involves two steps. First, the reaction mechanism is parsed and the relevant kinetic or thermodynamic information is stored in intermediate Kinetic Model and Aerosol Model data objects. These data are stored in dual Kokkos views, which allow synchronization of data between host and device memory. The data are further categorized and organized to optimize device-side kernel computation and facilitate postprocessing. For instance, for reactions following the Arrhenius form, TChem-atm stores both the reaction type and the associated parameters in separate dual views, as presented in Fig. 3.
Second, TChem-atm extracts only the data required by the computational kernels and synchronizes this subset to the device. The resulting constant data object – either kmcd or amcd – is then passed to the computation kernel. As a constant object, it cannot be modified during computation.
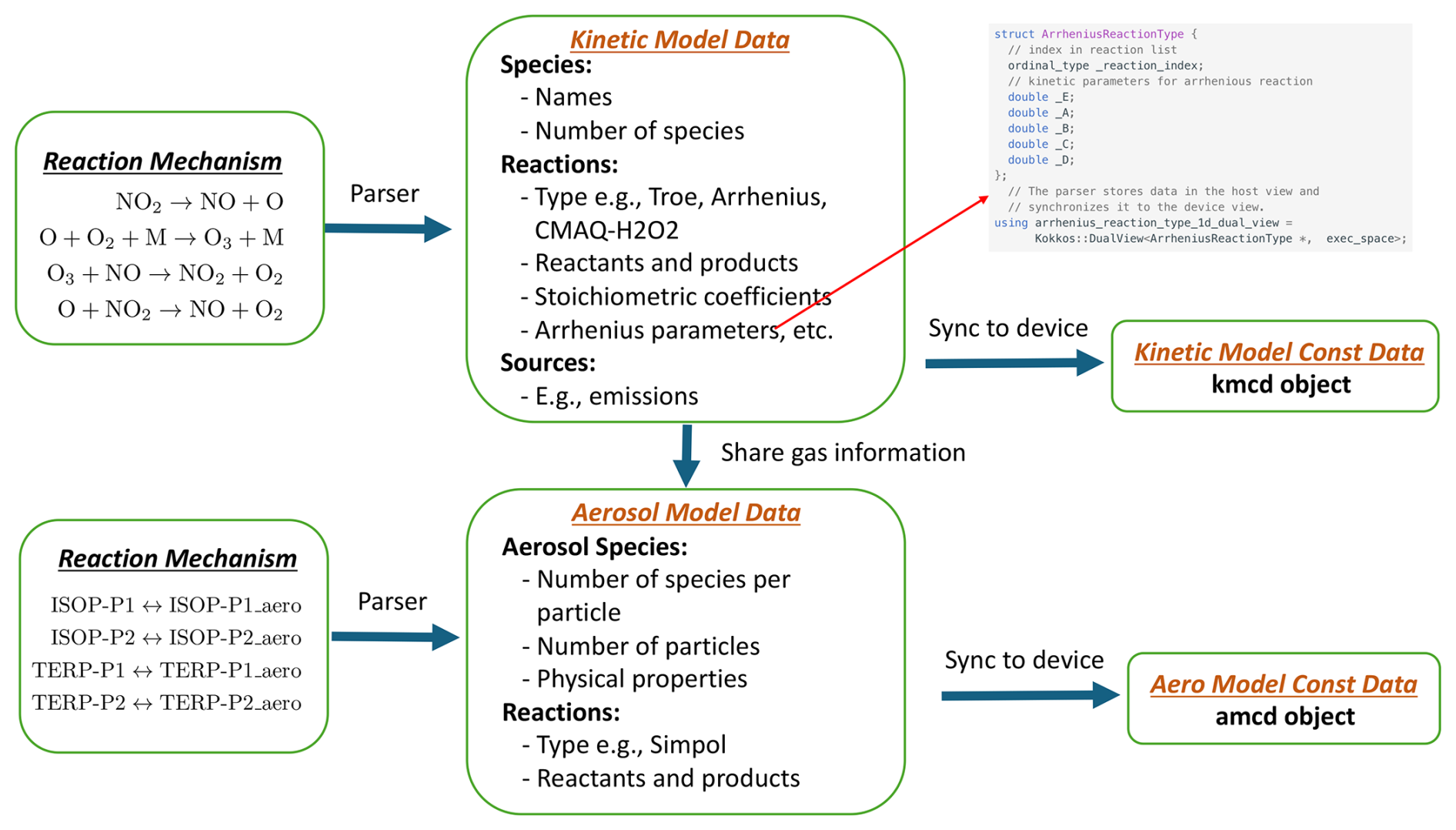
Figure 3TChem-atm's internal representation of the chemical mechanism is the kinetic model object.
2.4 System state vector and governing equations
We define the system state vector η as the concatenation of the gas-phase state vector g and aerosol-phase chemical state vector μ such that . The gas-phase state is represented by mixing ratios of K chemical species , while the aerosol phase is described by a set of N aerosol-phase vectors . Each aerosol-phase vector μi can be thought of as a “computational particle” i and contains the mass of A tracked species, .
In this section, we assume a particle-resolved context for illustrative purposes, where each μi corresponds to a single particle. However, TChem-atm can also be applied to sectional and modal models, where the computational particles are then mapped to sections and representative particles in a mode. A detailed discussion of this mapping is provided in Sect. 2.7.
In expanded form, the system state vector η is
In addition to the chemical state vector η, TChem-atm also requires the number concentration associated with each computational particle, denoted ni for particle i. This number concentration is not part of the state vector because it is not modified within TChem-atm. Instead, it is passed in by the host model and may evolve independently due to physical processes such as coagulation, nucleation, or sedimentation. Nonetheless, ni plays a critical role in the calculation of aerosol chemical source terms and is required for constructing the coupled gas–aerosol source terms described below.
The governing equations for the gas-phase and aerosol-phase components of the state vector are given by:
The source term Fgg,k accounts for gas-phase chemical reactions that produce or consume species k, as specified by the gas-phase chemical mechanism and further explained in Sect. 2.5. The term Fga,k represents gas–aerosol coupling processes, such as condensation of low-volatility species, evaporation of semi-volatile compounds, or uptake via heterogeneous reactions, and thus transfers mass between the gas phase and the aerosol phase. The corresponding aerosol-phase term, , describes the gain or loss of species a in computational particle i due to these interactions. These coupling terms ensure mass conservation across phases and encode the dynamic exchange of chemical species between gas and aerosol reservoirs and are explained in detail in Sect. 2.6. TChem-atm integrates the governing equations forward in time using the TINES or SUNDIALS CVODE ODE solver.
2.5 Gas Chemical Mechanism Source Term Construction
The gas-phase chemical source term, Fgg,k in Eq. (2), is computed via:
Here qj is the reaction rate of reaction j, , are stoichiometric coefficients for species k in reaction j, and Nreact is the total number of gas-phase reactions. The reaction rate is given by:
where kf,j is the reaction rate constant for reaction j. The functional forms for kf,j depend on reaction type, as listed in Table 2.
Currently, TChem-atm can reproduce gas chemistry for two complex reaction mechanisms: the gas chemistry of E3SM v3, i.e., the UCI chemistry system (University of California Irvine), and the Carbon Bond 2005 chemical mechanism, which is well-formulated for urban to remote tropospheric conditions (Yarwood et al., 2005). Because TChem-atm is mechanism-agnostic, it can be easily adapted to support a wide range of applications. For example, a minimal chemical mechanism can be defined for instructional use in classroom settings, while detailed mechanisms with hundreds of species and reactions can be incorporated for high-fidelity research simulations.
Table 2Gas reaction types available in TChem-atm; further details can be found in the TChem-atm online documentation.

2.6 Aerosol Mechanism Source Term Construction
TChem-atm computes the aerosol-gas interaction source terms, Fga,k and , as follows. In this section, we focus on the partitioning of semi-volatile gases. For simplicity, we assume that the first Nproc entries in the gas-phase species list correspond to partitioning species, and that each of these has a matching aerosol-phase counterpart with the same index a. That is, gas species ga partitions to aerosol species for .
where (s−1) is the mass transfer coefficient for species a and particle i, (mole mole−1) is the equilibrium mixing ratio of species a, ρa (kg m−3) is the mass density of gas species a needed for the conversion of mixing ratio to mass concentration, and ni is the number concentration of computational particle i needed to convert mass concentration to mass. While Fga,a is the mixing ratio flux of species a to the gas phase from all particles, is the mass flux of species a to computational particle i.
The mass transfer coefficient and the mass density ρa are given by,
where ri is the effective radius of the particle, is the gas-phase diffusion coefficient of species a, and is the Fuchs-Sutugin correction factor, p is atmospheric pressure, Ma is the molar weight of gas a (kg mole−1), R is the universal gas constant (J mole−1 K−1), and T is temperature (K).
The effective radius ri can be derived from the particle masses and aerosol species densities by assuming sphericity:
The Fuchs-Sutugin correction factor is computed as:
where is the Knudsen number, λa is the mean free path of gas a, and αa is the mass accommodation coefficient of gas a, here assumed to be 0.1 for all gases.
To calculate the equilibrium mixing ratios g* we use the SIMPOL.1 method (Pankow and Asher, 2008), which parameterizes as follows:
where is the equilibrium vapor pressure for gas a (Pa), p0=101 325.0 Pa is standard atmospheric pressure and , , , are fitting parameters provided by SIMPOL.1.
This section focuses on semi-volatile partitioning because this is the only heterogeneous process type currently implemented in TChem-atm. The framework, however, is extensible: heterogeneous reactions, reactive uptake parameterizations, or condensed-phase chemical processes can be incorporated by defining the corresponding rate expressions in the mechanism file and adding the associated source-term kernels. These capabilities follow the CAMP abstraction and will be implemented in future extensions of TChem-atm. At present, semi-volatile gas–aerosol partitioning represents the primary heterogeneous pathway supported.
2.7 Abstracting Aerosol Representations Across Models
One of CAMP's key innovations is to perform chemistry on individual computational particles, providing a unified library that works across different aerosol representations. As previously explained, a computational particle may represent an actual particle (in particle-resolved models), a bin section (in sectional models), or a representative particle within a mode (in modal models). This approach replaces the traditional coupling of chemistry to specific aerosol schemes – which is often hard-coded and inflexible – with a more general method that simplifies the addition of new species or processes. While this idea was originally described as an “abstraction of aerosol representation” in Dawson et al. (2022), the current framing in terms of computational particles offers a more intuitive and practical perspective, and steps toward the generalized representations envisioned in Hodzic et al. (2023).
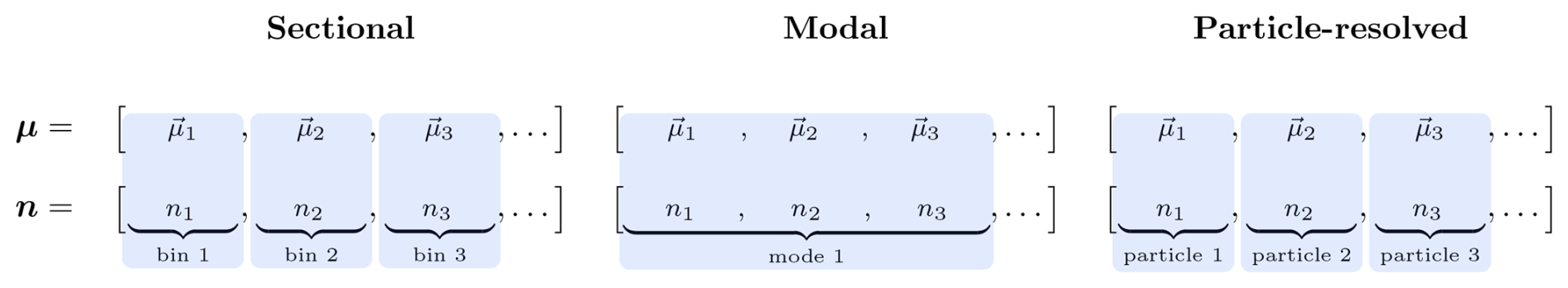
Figure 4Representation of the mapping between the abstract representation of the aerosol state vector μ in TChem-atm and the host model representation (blue filled regions). The host model is responsible for tracking the number concentration of each computational particle, contained in the n vector. Particle diameters are diagnosed from μ and n.
Following this approach, TChem-atm defines a general interface for aerosol–gas chemical interactions. Rather than prescribing how aerosols must be represented, TChem-atm relies on the host model to provide two essential pieces of information for each computational particle: (1) the chemical composition (i.e., the vector of aerosol species masses), and (2) the associated number concentration. As noted in Sect. 2.4, the number concentration is not part of the TChem-atm state vector, since it is not modified by the chemistry, but it plays a critical role in constructing source terms and must be supplied by the host model. Similarly, the particle diameter is required for processes such as surface-area-based uptake and size-dependent reaction rates. It is diagnosed from the particle masses and the number concentrations (assuming spherical particles). In particle-resolved models, this diameter is simply the diameter of the individual computational particle. In sectional models, it is typically taken as the midpoint of the bin.
For modal models, however, the situation is more complex. Modal schemes typically do not pass explicit diameters to the chemistry module; instead, they compute the effects of condensation by integrating over assumed size distributions and updating moments analytically – an approach rooted in the work of Whitby and McMurry (1997). While this method is widely used, it complicates the integration with approaches like TChem-atm that expect a well-defined particle diameter for each computational particle. This limitation is not a fundamental barrier, but addressing it would require additional work. Specifically, modal host models could be adapted to pass representative diameters (e.g., mean or characteristic values), which would improve compatibility with TChem-atm and enhance transparency and efficiency in multiphase chemistry calculations.
TChem-atm requires that each computational particle contains the full set of aerosol-phase species defined by the chemical mechanism. While individual species masses may be zero in any given computational particle, all species must be present in the data structure. This design ensures a consistent state vector structure across computational particles and simplifies source term construction. As a result, TChem-atm does not currently support mode-specific species sets, such as those used in some modal models where particular modes are restricted to subsets of species (e.g., black carbon only, or sea salt with organic carbon only). Any such mapping or aggregation must be handled externally by the host model prior to passing data to TChem-atm.
The abstractions within TChem-atm also facilitate integration with emerging or hybrid aerosol representations, such as adaptive sectional models, quadrature-based methods, or machine-learning-driven representations, as long as the host model can supply the required species masses, number concentrations, and diameters for each computational particle. Figure 4 illustrates how computational particles in TChem-atm correspond to different host model representations. For sectional and particle-resolved methods, there is a one-to-one mapping between computational particles tracked by TChem-atm and the corresponding host representation. For the modal approach, we display a scenario in which each mode is represented by a set of three characteristic computational particles with diameters corresponding to the geometric mean diameter and one standard deviation above and below.
2.8 Performance Portability with Kokkos
To enable performance portability across diverse computing platforms, PartMC-TChem-atm leverages the Kokkos library (Edwards et al., 2014; Trott et al., 2021, 2022). Kokkos is a programming model designed to abstract parallel execution and memory management, allowing a single implementation of numerical algorithms to run efficiently on a variety of architectures, including multi-core CPUs (OpenMP), NVIDIA GPUs (CUDA), AMD GPUs (HIP), and Intel GPUs (SYCL). Kokkos achieves this through compile-time polymorphism and execution policies that separate algorithm logic from hardware-specific optimizations. This abstraction is critical for modern atmospheric modeling, where simulations must scale across heterogeneous high-performance computing (HPC) systems.
Kokkos has been adopted in several modeling efforts to modernize physics and chemistry components for exascale computing. It underpins performance-portable implementations in the Energy Exascale Earth System Model (E3SM) atmosphere component (Taylor et al., 2023) and serves as the parallel backend for TChem (Kim et al., 2023). By integrating TChem-atm with Kokkos, PartMC-TChem-atm inherits the ability to execute detailed multiphase chemistry on both CPU and GPU architectures without modifying core algorithms.
2.9 Software Architecture and Jacobian Computation
TChem-atm adopts the same software design as the TChem library (Kim et al., 2023) and uses batched computations and algorithm implementations based on two primary Kokkos abstractions, ValueType and DeviceType. ValueType represents built-in scalar data types such as int and float, but it can also use automatic differentiation (AD) types from, for example, the SACADO package to support automatic computation of Jacobians and to facilitate differentiability for machine learning. DeviceType defines a memory space (where data lives, e.g., on CPU or GPU) and an execution space (where kernels run, e.g., on CPU or GPU). Each Kokkos backend supports a specific DeviceType so that applications written in Kokkos, using these abstractions, can run on diverse architectures. The workflow of TChem-atm is shown in Algorithm 1. It creates two data objects for gas and aerosol chemistry and uses hierarchical parallelism to batch-evaluate source terms or initial value problems. Nested team-thread parallelism enables performance across varying system configurations.
TChem-atm automatically computes Jacobian matrices using either finite difference methods or automatic differentiation via SACADO (Phipps et al., 2020). When ValueType is double, a numerical Jacobian is computed; TINES supports forward differencing, central differencing, and Richardson extrapolation, with forward differencing used by default. For analytical Jacobians, ValueType is an automatic differentiation type, and SACADO computes derivatives using the chain rule. This unified design simplifies the interface for downstream solvers.
As previously discussed, TChem-atm leverages Kokkos' team parallelism, utilizing batched computation. Specifically, multiple evaluations of nested routines to invoke a team function. The number of batches or samples is directly correlated with the number of grid points, as TChem-atm is invoked by an atmospheric model, or with model evaluations, where TChem-atm is utilized for uncertainty quantification and machine learning studies. Within the batched loop, a team routine is executed, with the primary team routine in TChem-atm being the right-hand-side routine, as shown in Algorithm 1.
For the gas component of the source term, nested parallel loops are employed to compute the kinetic constants for each reaction type (see Table 2), the rate of progress for each reaction, and the net production rate. For the aerosol component, nested parallel loops are similarly utilized for the evaluation of each aerosol process, where the number of particles or sections determines the size of the loop; currently, the SIMPOL process (Pankow and Asher, 2008) is supported within TChem-atm. Due to this batched design, we do not anticipate superior computational performance from TChem-atm when only a single evaluation is conducted. However, enhanced performance is expected as the number of evaluations increases.
Algorithm 1Batched algorithm for TChem-atm computation of chemical source terms.

2.10 ODE Solvers and Integration Workflow
TChem-atm is designed to decouple the specification of chemical processes from the numerical integration of the resulting ODE systems. In atmospheric models, gas- and aerosol-phase chemistry are typically solved independently at each grid point, often requiring the integration of stiff ODE systems over short time scales.
To support this need, TChem-atm provides interfaces to two widely used implicit solvers: TINES (Kim and Diaz-Ibarra, 2021) and SUNDIALS CVODE (Gardner et al., 2022; Hindmarsh et al., 2005; Balos et al., 2021; Hindmarsh et al., 2025), both of which implement backward differentiation formula (BDF) methods suitable for stiff systems. TINES employs a second-order Trapezoidal BDF (TrBDF2) scheme (Bank et al., 1985) with a Newton-based nonlinear solver and UTV decomposition for solving linear systems. CVODE supports both dense LU-based solvers, which require a Jacobian matrix and can use either analytical Jacobians (computed via automatic differentiation with SACADO (Phipps et al., 2020)) or numerical Jacobians (via finite difference schemes) (Salane, 1986), and GMRES-based iterative solvers, which do not require explicit Jacobians and instead approximate the action of the Jacobian through matrix-free methods.
To enable scalable integration across many independent ODE systems, TChem-atm adopts a batched interface, where source term evaluations and Jacobian computations are executed in parallel using Kokkos' team-level parallelism. This structure is compatible with large-scale simulations that solve thousands to millions of ODE systems simultaneously, such as grid-point chemical updates in 3D models.
Users can configure solver settings – including absolute and relative tolerances, maximum time steps, and method-specific options – via human-readable input files. This flexibility, along with modular interfaces to both solvers, allows users to tailor solver behavior to problem stiffness and computational constraints without modifying the underlying chemical mechanism or solver logic.
The interchangeable solver design also facilitates future extensibility. Additional solvers can be integrated with minimal code changes, supporting experimentation with different numerical strategies or coupling to new host models. Together, these features make TChem-atm a flexible and performance-portable platform for integrating chemically detailed, stiff ODE systems in a wide range of atmospheric chemistry applications. Performance comparisons across solver configurations – including dense and Jacobian-free methods on CPU and GPU architectures – are discussed in Sect. 3.2.
2.11 Testing Strategy
Development of TChem-atm is maintained via a GitHub repository. To ensure changes to the code base do not break model execution, numerous tests are run via GitHub Actions (GitHub, 2025) upon both the opening and merging of pull requests. Tests include small-scale unit testing of reaction and production rates, as well as larger-scale integration tests in which the entire chemical mechanism including E3SM's UCI chemistry system and the Carbon Bond 05 mechanism are evaluated. Additionally, construction of the source term is tested for the SIMPOL gas-aerosol system (Pankow and Asher, 2008) which describes partitioning of organic species.
2.12 Enabling Particle-Resolved GPU Chemistry
We integrated TChem-atm into the PartMC particle-resolved model to enable GPU-accelerated, chemically detailed simulations of multiphase aerosol chemistry. PartMC is a stochastic, particle-resolved aerosol box model that resolves the composition of many individual aerosol particles within a well-mixed volume of air. Riemer et al. (2009), DeVille et al. (2011), Curtis et al. (2016), and DeVille et al. (2019) describe in detail the numerical methods used in PartMC. PartMC tracks a large number of discrete computational particles to represent the aerosol population. Each computational particle is a vector of composition, which tracks the mass of each species within the particle and evolves the composition in time. By coupling PartMC with TChem-atm, each particle's chemistry is advanced using TChem-atm's batched source term evaluations and solver interface. Kokkos ensures that these evaluations are offloaded to GPUs with minimal overhead.
This integration allows PartMC to take advantage of the performance portability and multiphase chemistry capabilities developed in this work. The interface supports flexible mapping between PartMC's particle state variables and TChem-atm's kinetic model structure, preserving modularity and extensibility. In test cases, we validated the consistency of results across CPU and GPU architectures.
This section presents two sets of results. All simulations underlying the results presented here are fully reproducible using the archived code and dataset described in Diaz-Ibarra et al. (2025a, b, c). Because the goal of this work is to establish and validate the TChem-atm framework, the tests presented here focus on controlled box-model scenarios that isolate chemical behavior and computational scaling. First, we evaluate the chemical accuracy of the new TChem-atm integration within PartMC by comparing its outputs against the established PartMC-CAMP model in a box model simulation (Sect. 3.1). This includes gas-phase chemistry and gas–aerosol partitioning for a previously publised scenario (Dawson et al., 2022). Second, we assess the computational performance of the TChem-based implementation across multiple hardware backends (CPUs and GPUs) and solver configurations. We report per-particle wall-clock timings for both full ODE solves and individual kernel components such as RHS and Jacobian evaluations. Together, these results demonstrate the correctness and scalability of PartMC–TChem-atm for atmospheric chemistry modeling.
For this study, PartMC-TChem-atm was compiled and tested using Kokkos 4.5.01 with support for CPUs and GPUs. Simulations were performed on the following computing architectures: an NVIDIA H100 GPU on DeltaAI at the National Center for Supercomputing Applications (NCSA), University of Illinois; an AMD MI250x GPU on Frontier at the Oak Ridge Leadership Computing Facility (OLCF); and an AMD EPYC 7763 CPU on Perlmutter at the National Energy Research Scientific Computing Center (NERSC). The build process used the CMake-based infrastructure provided by TChem-atm, with Kokkos execution and memory spaces selected at compile time to target the appropriate backend. GPU simulations used the CUDA and HIP backends, while CPU runs used the OpenMP backend. All simulations used identical chemical mechanisms, numerical tolerances, and time step settings to isolate hardware performance and consistency of results.
3.1 PartMC-TChem-atm box model setup and comparison with PartMC-CAMP
To evaluate the performance of TChem-atm, we conducted a box model simulation using PartMC. The model was configured to represent a well-mixed air parcel with constant temperature and pressure. This simulation focuses exclusively on gas-phase chemistry and gas–aerosol partitioning, with coagulation and removal processes disabled. Gas-phase reactions were governed by the CB05 mechanism (Yarwood et al., 2005), and gas–aerosol partitioning followed the SIMPOL scheme (Pankow and Asher, 2008). The model was run for 24 h with a time step of 60 s. Initial gas-phase concentrations and emissions were set according to Table 10 of Dawson et al. (2022), while the aerosol phase was initialized with three modes, as described in the same reference. For the simulations presented here, we initialized PartMC with 103 computational particles. The complete set of code, model configuration files, initial conditions, and post-processing scripts used for this experiment are archived and publicly available (Diaz-Ibarra et al., 2025a, b, c).
Simulation results are shown in Fig. 5. Panel (a) displays the 24 h time series of ozone mixing ratios as predicted by PartMC-CAMP, PartMC-TChem-atm (CPU), and PartMC-TChem-atm (GPU). Panels (b)–(d) shows the evolution of gas-phase isoprene (ISOP) and its semi-volatile products, ISOP-P1 and ISOP-P2. ISOP-P1 is formed through the reaction of ISOP with OH, while ISOP-P2 results from its reaction with ozone. Figure 5e–f presents the aerosol-phase mass concentrations of ISOP-P1_aero and ISOP-P2_aero, which arise from the partitioning of their respective gas-phase precursors.
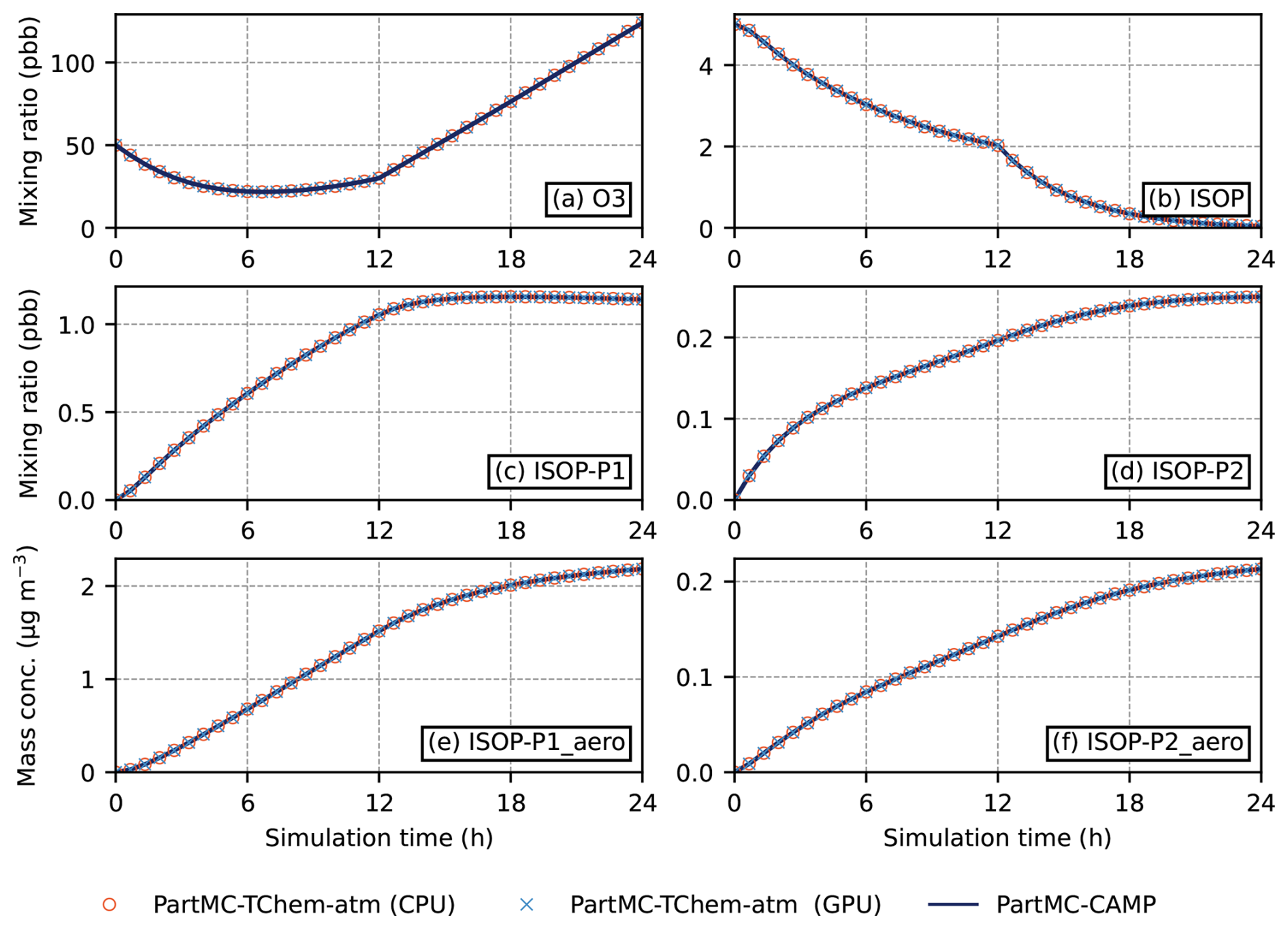
Figure 5Comparison between PartMC-TChem-atm (CPU), PartMC-TChem-atm (GPU) and PartMC-CAMP for (a) ozone mixing ratio, (b) ISOP, (c) ISOP-P1 and (d) ISOP-P2 mixing ratios, (e) ISOP-P1_aero and (f) ISOP-P2_aero mass concentration for the 24 h simulation period.
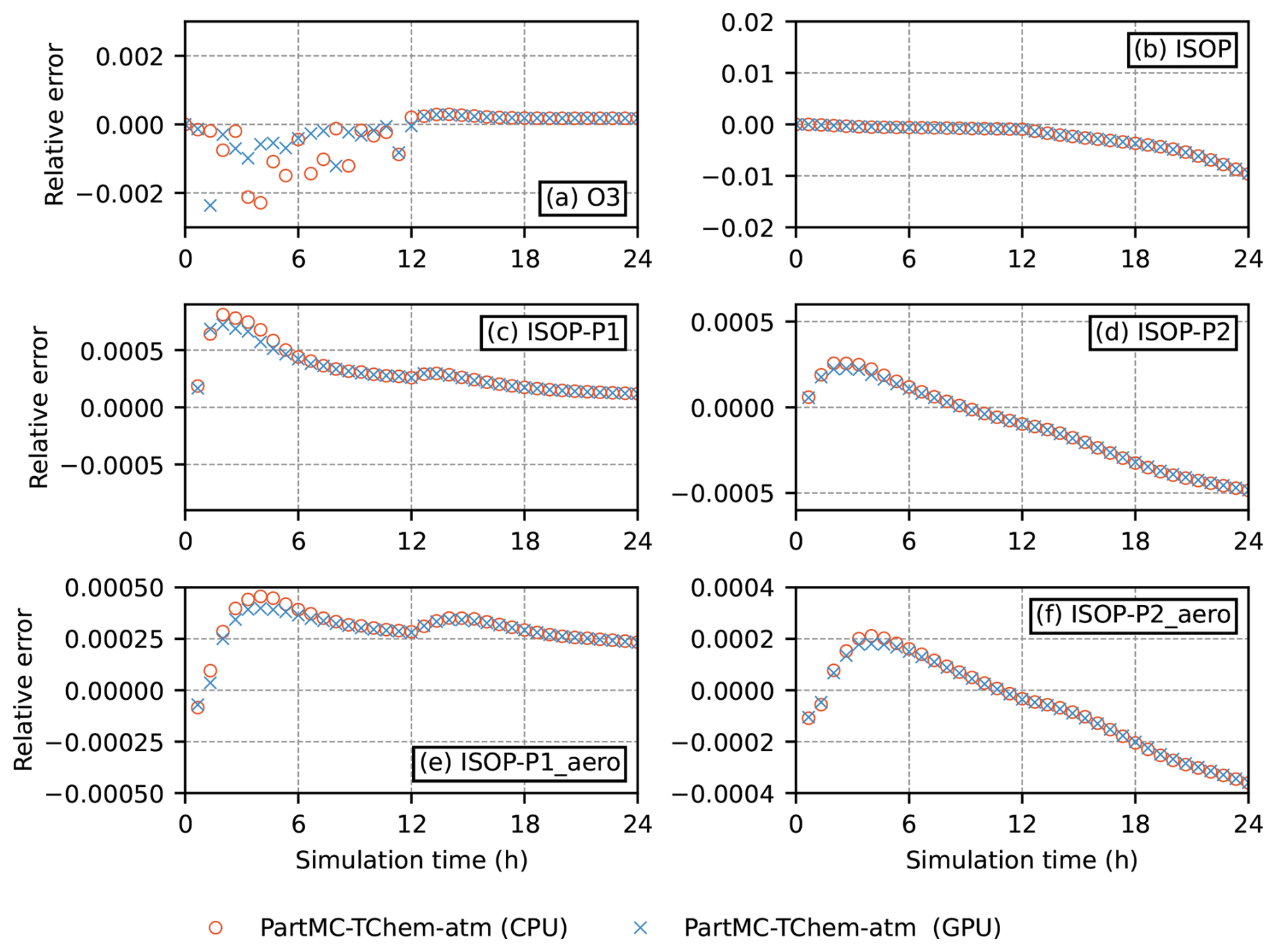
Per-timestep relative tolerances provided by TChem-atm to the CVODE solver control the final accumulated error. In order to achieve 0.01 % global accuracy for this comparison, a relative tolerance of 10−6 was chosen. To summarize how PartMC-TChem-atm compares to PartMC-CAMP, the ℓ2-norm of the relative difference was for gas phase species and for aerosol species.
To more clearly quantify the agreement between the CPU, GPU, and legacy PartMC–CAMP implementations, Fig. 6 shows the relative differences between the CPU, GPU, and PartMC–CAMP results. The discrepancies remain small throughout the simulation, with a modest temporal drift for some species that reflects normal numerical variability rather than any architecture-specific effect. All deviations remain well within solver tolerances.
Overall, PartMC-TChem-atm reproduces the results of PartMC-CAMP with high fidelity. It performs consistently across both CPU and GPU architectures, capturing expected chemical behavior while enabling accelerated simulations on heterogeneous computing platforms.
3.2 Benchmarking GPU Speedup and Solver Scaling
We evaluate the computational performance of TChem-atm on several hardware backends including multicore CPUs and both NVIDIA and AMD GPU architectures. To achieve this, the box model example from Sect. 3.1 was run by varying the number of computational particles, which increases the size of the source term and the Jacobian matrix. We tested several ODE solver configurations presented in Table 3, including TINES TrBDF2 and two variants of SUNDIALS CVODE that differ by the sparsity of the linear solver (SUNDIALS CVODE-GMRES and SUNDIALS CVODE (dense)). The same numerical parameters are applied for the three configurations.
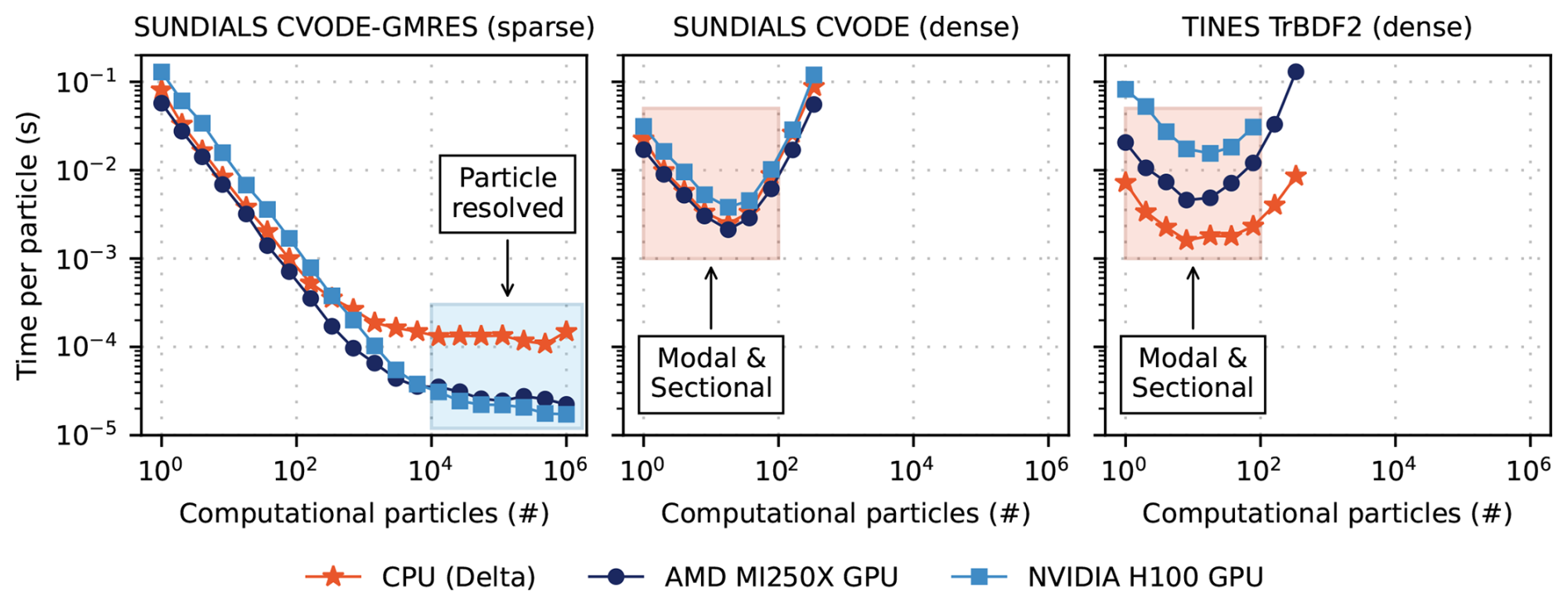
Figure 7Per-particle solver wall clock time through t=10 s organized by solver. Timings are colored by computing architecture. For each solver and architecture, team size and vector size were set via the Kokkos::AUTO configuration. TINES TrBDF2 timings for the AMD MI250X at particle counts larger than Np∼100 are not included due to the required memory exceeding allocatable GPU DRAM capacity. Shaded regions indicate approximate computing regimes for each aerosol treatment.
For all numerical experiments, the model was run to a simulated time of 10 s and the wall clock time per time step was measured. For each solver, the number of computational particles was varied from Np=1 to in twenty, logarithmically spaced increments. Figure 7 shows the wall clock time per particle for each solver configuration across a range of particle counts. For small particle numbers (i.e., Np≲50), solvers using dense linear algebra – namely SUNDIALS CVODE (dense) and TINES TrBDF2 – achieve the best performance due to their low overhead and efficient handling of small Jacobians. However, as Np increases, the cost of forming and solving dense Jacobian systems grows rapidly, making these approaches computationally prohibitive beyond Np∼500.
In contrast, SUNDIALS CVODE-GMRES employs a Jacobian-free Krylov method, which avoids explicit construction of the Jacobian. This enables continued scaling to larger particle numbers, with wall clock time per particle decreasing even up to Np=106 on GPU architectures. These results underscore the importance of matching solver structure to problem size and hardware – dense solvers excel at small scale, while Jacobian-free methods offer superior scalability.
In the GMRES case, we observe substantial speedups of GPU relative to CPU at large computational particle numbers, underscoring the advantages of Jacobian-free Krylov methods on GPUs. These results are based on a single-cell configuration, however, and therefore do not yet fully exploit the parallelism available on GPU architectures. In larger multi-cell simulations, we anticipate even greater gains for GMRES as well as improvements for dense solvers, which are not fully reflected under the present single-cell conditions.

These results have practical implications for model selection and hardware deployment. Particle-resolved simulations such as PartMC typically require on the order of 10 000 computational particles per grid box to adequately resolve mixing state and aerosol composition. In this regime, dense linear solvers become inefficient, and the GPU implementation of SUNDIALS CVODE-GMRES offers significant performance advantages due to its Jacobian-free formulation and better scalability with particle number.
Conversely, if the application involves simplified aerosol representations – such as modal or sectional models with a small number of size/composition bins – the computational burden is much lower, and CPU-based solvers with dense linear algebra may offer better performance. Thus, the optimal hardware–solver combination depends strongly on the complexity and resolution of the aerosol representation. To further illustrate the relationship between aerosol treatment and the choice of solver, approximate computing regimes for each aerosol treatment are shown as shaded regions in Fig. 7.
Timings shown in Fig. 7 were conducted using the Kokkos:AUTO configuration, which automatically sets the level of parallelism at both the team and vector level (for those familiar with NVIDIA systems, the Kokkos team size and vector size are cast as the x and y dimension of each CUDA block). Both parameters can be set at runtime to determine a problem-specific optimal configuration for team and vector size. Figure 8 shows heatmaps of wall clock time per particle for the SUNDIALS CVODE-GMRES solver where both the team size and vector size are varied. Optimal team and vector size configurations differ by GPU. For the NVIDIA H100, a team size of 16 and a vector length of 32 resulted in the lowest wall clock time per particle, whereas a team size of 64 and vector length of 16 were best for the AMD MI250X. Below each heatmap in Fig. 8 are the minimum, maximum, and mean wall clock time across each team and vector configuration alongside the wall clock time achieved by the Kokkos:AUTO configuration. We find that the optimal configuration for the NVIDIA H100 is within a factor of 2 faster than the Kokkos:AUTO configuration, while both are equal for the AMD MI250X.
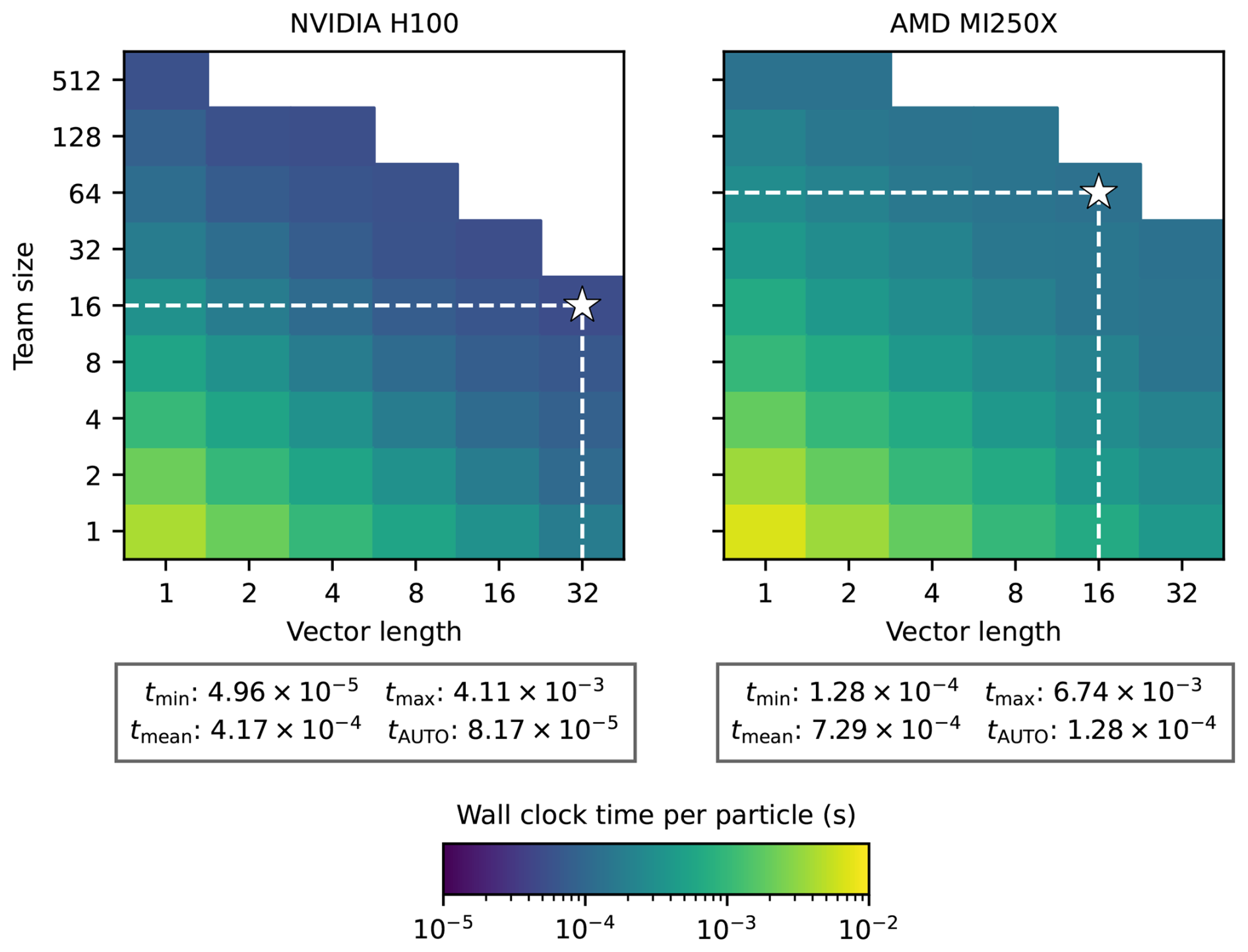
Figure 8Heatmaps of wall clock time per particle as a function of Kokkos team size and vector length for the SUNDIALS CVODE-GMRES solver on the NVIDIA H100 (left) and AMD MI250X (right). For each solver run, the number of particles was fixed at 1000. The optimal configuration (lowest wall clock time) is indicated by a white star in each plot.
Ultimately, solver performance is governed by the size and structure of the Jacobian matrix. A large number of particles or reactions leads to a large Jacobian, while a large number of chemical species often results in a sparse one. The balance between these factors – reaction count, species count, and particle/bin resolution – ultimately determines whether dense or iterative solvers, and CPU or GPU hardware, offer the most efficient path forward.
In addition to full ODE solver runs, a separate set of timings were conducted to evaluate the performance of the RHS evaluation and Jacobian construction. Figure 9 shows how wall clock time for the RHS and Jacobian vary as the number of particles increases.
For each computing backend, per-particle performance for RHS evaluations improves as the number of particles increases. When , we find the best performance for the NVIDIA H100. Above , the AMD MI250X achieves the best performance through . At high particle counts (), performance plateaus for each computing backend. At , GPU backends achieve speedups of 2.3 and 5 relative to the CPU for the NVIDIA H100 and AMD MI250X, respectively.
Additionally, Jacobian evaluation timings exhibit similar performance for both the CPU and GPUs: wall clock time per particle decreases when the number of particles is low (i.e., Np≲100), however, performance degrades as Np increases further due to corresponding growth in the Jacobian.
To interpret these performance trends, it is helpful to clarify the role of direct versus iterative solvers. We include dense-direct linear solvers in our performance evaluation not because they are expected to be optimal for large atmospheric chemistry systems – indeed, the Jacobians arising from CB05 and similar mechanisms are sparse – but because they provide a well-defined performance baseline for small to moderate system sizes. Dense solvers incur higher memory costs and scale poorly as the number of particles grows, so their performance degradation is expected. However, these solvers remain competitive in regimes with few computational particles or simplified aerosol representations (e.g., small sectional or modal systems), where the Jacobian dimension remains modest and overhead is low. Including them therefore helps delineate the transition point at which preconditioned iterative methods such as GMRES become the clearly superior choice. Our intention is not to advocate dense solvers for large, stiff atmospheric chemistry problems, but rather to provide a complete performance map across solver categories and hardware backends. For realistically sized atmospheric mechanisms, preconditioned iterative Krylov methods are preferable because they exploit sparsity and dramatically reduce memory demands. The results in Sect. 3.2 are therefore consistent with expectations and are included to contextualize solver performance across operational regimes.
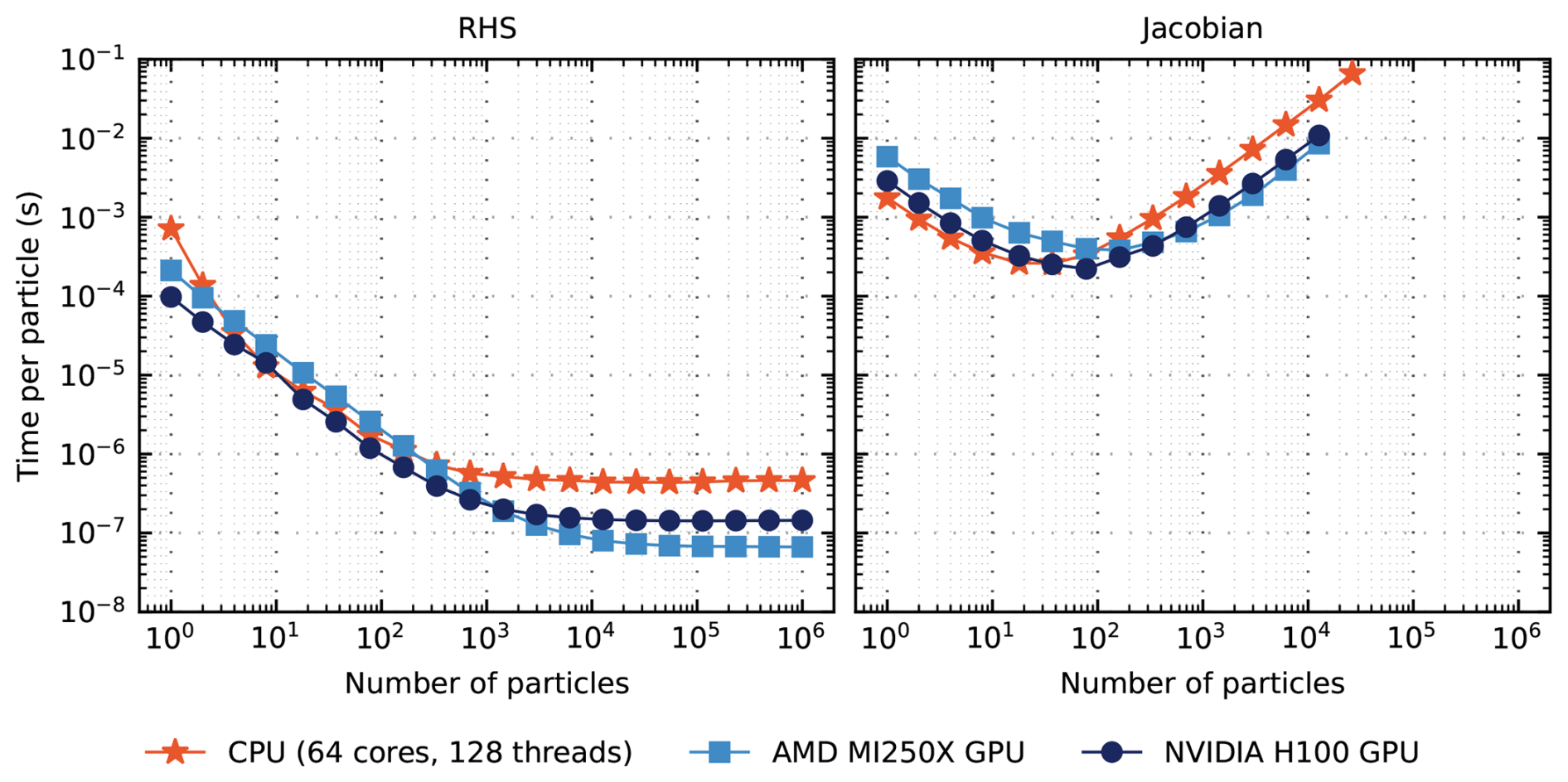
Figure 9Per-particle wall clock time for evaluation of the gas-aerosol right-hand side (left) and Jacobian (right). Timings are colored by computing architecture. For each timing run, team size and vector size were set via the Kokkos::AUTO configuration. Particle number is limited for Jacobian evaluations due to exceeding the allocatable memory of each computing architecture.
We also conducted lightweight profiling during the timing experiments to separate kernel execution from host–device memory transfer. For the idealized box-model runs presented here, more than 99.9 % of the wall-clock time occurs inside the Kokkos kernels, with memory transfers accounting for only the remaining fraction. This behavior is expected in the present setup, where particles share identical initial conditions, the RHS evaluation is uniform across particles, and memory movement is minimized. In more realistic host-model applications, such as full PartMC–TChem-atm simulations, additional data exchange between the host model and TChem-atm, more heterogeneous particle states, and increased stiffness may increase the relative cost of memory transfers and solver iterations. Thus, while our profiling confirms that kernel execution dominates in these proof-of-concept tests, the performance breakdown will necessarily depend on the host-model coupling pattern and the chemical and microphysical variability of the simulated system.
3.3 Potential Applications in Chemistry–Transport and Climate Models
While the tests presented in this paper focus on idealized box-model configurations to isolate numerical behavior and performance, TChem-atm is designed for use in full chemistry–transport and Earth system models. CAMP, from which TChem-atm adopts its multiphase abstractions, has already been integrated into the MONARCH model system (Dawson et al., 2022), demonstrating the suitability of the CAMP structure for real-world chemical forecasting and case-study applications. To date, TChem-atm has not yet been deployed in operational chemistry–transport models; however, the TChem-atm interface is compatible with the operator-splitting approach used in such models, where chemistry is solved independently in each grid cell as a box model. Integration therefore requires only the development of a host-model-specific interface, not changes to TChem-atm itself.
A near-term target for deployment is the Energy Exascale Earth System Model (E3SM), where the Kokkos-based design of TChem-atm is well aligned with ongoing efforts to modernize the atmospheric chemistry infrastructure for heterogeneous computing environments. Exploring this integration constitutes part of our future work and leverages the fact that TChem-atm already implements the data structures and parallelism patterns required for grid-level chemistry calls in a chemistry–transport or climate model.
We have presented TChem-atm, a performance-portable approach for simulating chemically detailed, multiphase atmospheric chemistry across diverse computing architectures. By integrating the chemical mechanism infrastructure of CAMP with the Kokkos-based backend of TChem, TChem-atm enables consistent deployment across CPUs and GPUs while maintaining flexibility in mechanism configuration and solver selection.
A key strength of TChem-atm is its support for multiple aerosol representations – including modal, sectional, and particle-resolved approaches – making it suitable for a wide range of host models and applications. Integration with the particle-resolved model PartMC demonstrates that TChem-atm accurately reproduces gas-phase chemistry and gas–aerosol partitioning results, verifying the method's correctness and interoperability.
Performance benchmarks reveal that dense linear solvers are well-suited for small particle populations, whereas Jacobian-free solvers such as CVODE-GMRES offer scalable performance benefits on GPUs as the number of computational particles increases. These gains are particularly significant for particle-resolved models like PartMC, which typically require around 10 000 particles per grid cell to capture aerosol mixing state. Across solvers, our modular approach supports efficient, scalable integration of detailed multiphase chemistry across diverse computing platforms.
While the current version of TChem-atm is not yet fully performance-optimized, it provides a solid and extensible foundation for future development. Ongoing efforts will focus on enhanced solver strategies, improved memory layout, and broader chemical mechanism support. The code and all associated test cases are publicly available under an open-source license, enabling verification, reuse, and extension by the broader atmospheric modeling community (Diaz-Ibarra et al., 2025a, b, c).
TChem-atm is open source and available via GitHub under the BSD 2-Clause License at: https://github.com/pcLAeroParams/TChem-atm/ (last access: 30 January 2026). The version of the code used in this study has been archived on Zenodo under https://doi.org/10.5281/zenodo.17058144 (Diaz-Ibarra et al., 2025a). TChem-atm depends on Kokkos, SUNDIALS, and TINES, which are also openly available. PartMC is open source and available via GitHub at https://github.com/compdyn/PartMC (last access: 30 January 2026) and Zenodo at https://doi.org/10.5281/zenodo.17309370 (Diaz-Ibarra et al., 2025c). All simulation inputs and output used to generate the figures in this paper are archived at https://doi.org/10.5281/zenodo.17362352 (Diaz-Ibarra et al., 2025b).
OD led the implementation and development of TChem-atm, coordinated computing resources. SGF developed code and performed benchmarking and scalability analysis (Sect. 3.2), curated data, coordinated computing resources. JHC led the integration of TChem-atm and PartMC, conducted simulations and analysis (Sect. 3.1) and curated data. ZD performed code review and technical validation. MW provided overall guidance. NR coordinated the project, outlined and organized the manuscript, and synthesized the final text. All authors contributed to editing.
The contact author has declared that none of the authors has any competing interests.
The employees co-own right, title and interest in and to the article and are responsible for its contents. The United States Government retains, and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this article or allow others to do so, for United States Government purposes. The DOE will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan at https://www.energy.gov/downloads/doe-public-access-plan (last access: 30 January 2026).
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. The authors bear the ultimate responsibility for providing appropriate place names. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
This work used computing resources from DeltaAI at the National Center for Supercomputing Applications (NCSA), University of Illinois Urbana-Champaign; Frontier at the Oak Ridge Leadership Computing Facility (OLCF); and Perlmutter at the National Energy Research Scientific Computing Center (NERSC). The authors thank the developers of Kokkos, TChem, TINES, and SUNDIALS for their contributions to the open-source ecosystem that made this work possible.
This work was supported by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research under Award Number DE-SC0022130, the National Science Foundation Grant No. AGS 19-41110, and the Laboratory Directed Research and Development program at Sandia National Laboratories. Sandia National Laboratories is a multimission laboratory managed and operated by National Technology and Engineering Solutions of Sandia LLC, a wholly owned subsidiary of Honeywell International Inc. for the U.S. Department of Energy’s National Nuclear Security Administration contract DE-NA0003525.
This paper was edited by Patrick Jöckel and reviewed by two anonymous referees.
Alvanos, M. and Christoudias, T.: GPU-accelerated atmospheric chemical kinetics in the ECHAM/MESSy (EMAC) Earth system model (version 2.52), Geoscientific Model Development, 10, 3679–3693, https://doi.org/10.5194/gmd-10-3679-2017, 2017. a
Appel, K. W., Bash, J. O., Fahey, K. M., Foley, K. M., Gilliam, R. C., Hogrefe, C., Hutzell, W. T., Kang, D., Mathur, R., Murphy, B. N., Napelenok, S. L., Nolte, C. G., Pleim, J. E., Pouliot, G. A., Pye, H. O. T., Ran, L., Roselle, S. J., Sarwar, G., Schwede, D. B., Sidi, F. I., Spero, T. L., and Wong, D. C.: The Community Multiscale Air Quality (CMAQ) model versions 5.3 and 5.3.1: system updates and evaluation, Geoscientific Model Development, 14, 2867–2897, https://doi.org/10.5194/gmd-14-2867-2021, 2021. a
Balos, C. J., Gardner, D. J., Woodward, C. S., and Reynolds, D. R.: Enabling GPU accelerated computing in the SUNDIALS time integration library, Parallel Computing, 108, 102836, https://doi.org/10.1016/j.parco.2021.102836, 2021. a, b
Bank, R. E., Coughran, W. M., Fichtner, W., Grosse, E. H., Rose, D. J., and Smith, R. K.: Transient Simulation of Silicon Devices and Circuits, IEEE Transactions on Computer-Aided Design of Integrated Circuits and Systems, 4, 436–451, 1985. a
Bey, I., Jacob, D. J., Yantosca, R. M., Logan, J. A., Field, R. B., Fiore, A. M., Li, Q., Liu, H., Mickley, L. J., and Schultz, M. G.: Global modeling of tropospheric chemistry with assimilated meteorology: Model description and evaluation, Journal of Geophysical Research: Atmospheres, 106, 23073–23095, https://doi.org/10.1029/2001JD000807, 2001. a
Byun, D. W. and Schere, K. L.: Review of the governing equations, computational algorithms, and other components of the Models-3 Community Multiscale Air Quality (CMAQ) modeling system, Applied Mechanics Reviews, 59, 51–77, https://doi.org/10.1115/1.2128636, 2006. a
Cao, K., Wu, Q., Wang, L., Wang, N., Cheng, H., Tang, X., Li, D., and Wang, L.: GPU-HADVPPM V1.0: a high-efficiency parallel GPU design of the piecewise parabolic method (PPM) for horizontal advection in an air quality model (CAMx V6.10), Geoscientific Model Development, 16, 4367–4383, https://doi.org/10.5194/gmd-16-4367-2023, 2023. a
Curtis, J. H., Michelotti, M. D., Riemer, N., Heath, M. T., and West, M.: Accelerated simulation of stochastic particle removal processes in particle-resolved aerosol models, J. Comput. Phys., 322, 21–32, 2016. a
Dawson, M. L., Guzman, C., Curtis, J. H., Acosta, M., Zhu, S., Dabdub, D., Conley, A., West, M., Riemer, N., and Jorba, O.: Chemistry Across Multiple Phases (CAMP) version 1.0: an integrated multiphase chemistry model, Geoscientific Model Development, 15, 3663–3689, https://doi.org/10.5194/gmd-15-3663-2022, 2022. a, b, c, d, e, f
DeVille, L., Riemer, N., and West, M.: Convergence of a generalized Weighted Flow Algorithm for stochastic particle coagulation, Journal of Computational Dynamics, 1–18, https://doi.org/10.3934/jcd.2019003, 2019. a
DeVille, R. E. L., Riemer, N., and West, M.: Weighted Flow Algorithms (WFA) for stochastic particle coagulation, J. Comput. Phys., 230, 8427–8451, 2011. a
Diaz-Ibarra, O. H., Frederick, S., Curtis, J., D'Aquino, Z., Safta, C., West, M., and Riemer, N.: TChem-atm version 2.0.0, Zenodo [code], https://doi.org/10.5281/zenodo.17058144, 2025a. a, b, c, d
Diaz-Ibarra, O. H., Frederick, S., Curtis, J., D'Aquino, Z., Bosler, P., Patel, L., Safta, C., West, M., and Riemer, N.: Data for TChem-atm (v2.0.0): Scalable Performance-Portable Multiphase Atmospheric Chemistry, Zenodo [data set], https://doi.org/10.5281/zenodo.17362352, 2025b. a, b, c, d
Diaz-Ibarra, O. H., Frederick, S., Curtis, J., D'Aquino, Z., Safta, C., West, M., and Riemer, N.: PartMC-TChem-atm, Zenodo [code], https://doi.org/10.5281/zenodo.17309370, 2025c. a, b, c, d
Eastham, S. D., Long, M. S., Keller, C. A., Lundgren, E., Yantosca, R. M., Zhuang, J., Li, C., Lee, C. J., Yannetti, M., Auer, B. M., Clune, T. L., Kouatchou, J., Putman, W. M., Thompson, M. A., Trayanov, A. L., Molod, A. M., Martin, R. V., and Jacob, D. J.: GEOS-Chem High Performance (GCHP v11-02c): a next-generation implementation of the GEOS-Chem chemical transport model for massively parallel applications, Geoscientific Model Development, 11, 2941–2953, https://doi.org/10.5194/gmd-11-2941-2018, 2018. a
Edwards, H. C., Trott, C. R., and Sunderland, D.: Kokkos: Enabling manycore performance portability through polymorphic memory access patterns, Journal of Parallel and Distributed Computing, 74, 3202–3216, 2014. a
Gardner, D. J., Reynolds, D. R., Woodward, C. S., and Balos, C. J.: Enabling new flexibility in the SUNDIALS suite of nonlinear and differential/algebraic equation solvers, ACM Transactions on Mathematical Software (TOMS), 48, 1–24, https://doi.org/10.1145/3539801, 2022. a, b
GitHub: GitHub Actions, https://github.com/features/actions (last access: 30 January 2026), 2025. a
Golaz, J. C., Caldwell, P. M., Van Roekel, L. P., Petersen, M. R., Tang, Q., Wolfe, J. D., Abeshu, G., Anantharaj, V., Asay-Davis, X. S., Bader, D. C., Baldwin, S. A., Bisht, G., Bogenschutz, P. A., Branstetter, M., Brunke, M. A., Brus, S. R., Burrows, S. M., Cameron-Smith, P. J., Donahue, A. S., Deakin, M., Easter, R. C., Evans, K. J., Feng, Y., Flanner, M., Foucar, J. G., Fyke, J. G., Griffin, B. M., Hannay, C., Harrop, B. E., Hoffman, M. J., Hunke, E. C., Jacob, R. L., Jacobsen, D. W., Jeffery, N., Jones, P. W., Keen, N. D., Klein, S. A., Larson, V. E., Leung, L. R., Li, H. Y., Lin, W. Y., Lipscomb, W. H., Ma, P. L., Mahajan, S., Maltrud, M. E., Mametjanov, A., McClean, J. L., McCoy, R. B., Neale, R. B., Price, S. F., Qian, Y., Rasch, P. J., Eyre, J. E. J. R., Riley, W. J., Ringler, T. D., Roberts, A. F., Roesler, E. L., Salinger, A. G., Shaheen, Z., Shi, X. Y., Singh, B., Tang, J. Y., Taylor, M. A., Thornton, P. E., Turner, A. K., Veneziani, M., Wan, H., Wang, H. L., Wang, S. L., Williams, D. N., Wolfram, P. J., Worley, P. H., Xie, S. C., Yang, Y., Yoon, J. H., Zelinka, M. D., Zender, C. S., Zeng, X. B., Zhang, C. Z., Zhang, K., Zhang, Y., Zheng, X., Zhou, T., and Zhu, Q.: The DOE E3SM Coupled Model Version 1: Overview and Evaluation at Standard Resolution, Journal of Advances in Modeling Earth Systems, 11, 2089–2129, https://doi.org/10.1029/2018MS001603, 2019. a
Hindmarsh, A. C., Brown, P. N., Grant, K. E., Lee, S. L., Serban, R., Shumaker, D. E., and Woodward, C. S.: SUNDIALS: Suite of nonlinear and differential/algebraic equation solvers, ACM Transactions on Mathematical Software (TOMS), 31, 363–396, https://doi.org/10.1145/1089014.1089020, 2005. a, b
Hindmarsh, A. C., Serban, R., Balos, C. J., Gardner, D. J., Reynolds, D. R., and Woodward, C. S.: User Documentation for CVODE, v7.4.0, https://sundials.readthedocs.io/en/latest/cvode (last access: 30 January 2026), 2025. a, b
Hodzic, A., Mahowald, N., Dawson, M., Johnson, J., Bernardet, L., Bosler, P. A., Fast, J. D., Fierce, L., Liu, X., Ma, P.-L., Murphy, B., Riemer, N., and Schulz, M.: Generalized Aerosol/Chemistry Interface (GIANT): A Community Effort to Advance Collaborative Science across Weather and Climate Models, Bulletin of the American Meteorological Society, 104, E2065–E2080, 2023. a, b
Kim, K. and Diaz-Ibarra, O.: Tines, Github, https://github.com/sandialabs/tines (last access: 11 May 2021), 2021. a, b
Kim, K., Díaz-Ibarra, O. H., Najm, H. N., Zádor, J., and Safta, C.: TChem: A performance portable parallel software toolkit for complex kinetic mechanisms, Computer Physics Communications, 285, 108628, https://doi.org/10.1016/j.cpc.2022.108628, 2023. a, b, c, d
Pankow, J. F. and Asher, W. E.: SIMPOL.1: a simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds, Atmospheric Chemistry and Physics, 8, 2773–2796, https://doi.org/10.5194/acp-8-2773-2008, 2008. a, b, c, d, e
Phipps, E. T., Pawlowski, R. P., and Trott, C. R.: Automatic Differentiation of C++ Codes on Emerging Manycore Architectures with Sacado, Tech. Rep. SAND2020-8010C, Sandia National Laboratories, https://doi.org/10.1145/3560262, 2020. a, b
Pye, H. O. T., Place, B. K., Murphy, B. N., Seltzer, K. M., D'Ambro, E. L., Allen, C., Piletic, I. R., Farrell, S., Schwantes, R. H., Coggon, M. M., Saunders, E., Xu, L., Sarwar, G., Hutzell, W. T., Foley, K. M., Pouliot, G., Bash, J., and Stockwell, W. R.: Linking gas, particulate, and toxic endpoints to air emissions in the Community Regional Atmospheric Chemistry Multiphase Mechanism (CRACMM), Atmospheric Chemistry and Physics, 23, 5043–5099, https://doi.org/10.5194/acp-23-5043-2023, 2023. a
Quevedo, D., Do, K., Delic, G., Rodríguez-Borbón, J., Wong, B. M., and Ivey, C. E.: GPU Implementation of a Gas-Phase Chemistry Solver in the CMAQ Chemical Transport Model, ACS ES&T Air, https://doi.org/10.1021/acsestair.4c00181, 2025. a
Rasch, P., Xie, S., Ma, P.-L., Lin, W., Wang, H., Tang, Q., Burrows, S., Caldwell, P., Zhang, K., Easter, R., Cameron-Smith, P., Singh, B., Wan, H., Golaz, J.-C., Harrop, B. E., Roesler, E., Bacmeister, J., Larson, V. E., Evans, K. J., Qian, Y., Taylor, M., Leung, L. R., Zhang, Y., Brent, L., Branstetter, M., Hannay, C., Mahajan, S., Mametjanov, A., Neale, R., Richter, J. H., Yoon, J.-H., Zender, C. S., Bader, D., Flanner, M., Foucar, J. G., Jacob, R., Keen, N., Klein, S. A., Liu, X., Salinger, A., Shrivastava, M., and Yang, Y.: An Overview of the Atmospheric Component of the Energy Exascale Earth System Model (E3SM), Journal of Advances in Modeling Earth Systems, 11, 2377–2410, https://doi.org/10.1029/2019MS001629, 2019. a
Riemer, N., West, M., Zaveri, R. A., and Easter, R. C.: Simulating the evolution of soot mixing state with a particle-resolved aerosol model, Journal of Geophysical Research: Atmospheres, 114, D09202, https://doi.org/10.1029/2008JD011073, 2009. a
Ruiz, C. G., Acosta, M., Jorba, O., Galobardes, E. C., Dawson, M., Oyarzun, G., García-Pando, C. P., and Serradell, K.: Optimized thread-block arrangement in a GPU implementation of a linear solver for atmospheric chemistry mechanisms, Computer Physics Communications, 302, 109240, https://doi.org/10.1016/j.cpc.2024.109240, 2024. a
Salane, D. E.: Adaptive routines for forming Jacobians numerically, SAND86, https://www.osti.gov/biblio/5271352 (last access: 30 January 2026), 1986. a
Sun, J., Fu, J. S., Drake, J. B., Zhu, Q., Haidar, A., Gates, M., Tomov, S., and Dongarra, J.: Computational benefit of GPU optimization for the atmospheric chemistry modeling, Journal of Advances in Modeling Earth Systems, 10, 1952–1969, 2018. a
Taylor, M., Caldwell, P. M., Bertagna, L., Clevenger, C., Donahue, A., Foucar, J., Guba, O., Hillman, B., Keen, N., Krishna, J., Norman, M., Sreepathi, S., Terai, C., White, J. B., Salinger, A. G., McCoy, R. B., Leung, L.-Y. R., Bader, D. C., and Wu, D.: The simple cloud-resolving E3SM atmosphere model running on the Frontier exascale system, in: Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, 1–11, 2023. a
Trott, C. R., Lebrun-Grandié, D., Arndt, D., Ciesko, J., Dang, V., Ellingwood, N., Gayatri, R., Harvey, E., Hollman, D. S., Ibanez, D., Liber, N., Madsen, J., Miles, J., Poliakoff, D., Powell, A., Rajamanickam, S., Simberg, M., Sunderland, D., Turcksin, B., and Wilke, J.: Kokkos 3: Programming model extensions for the exascale era, IEEE Transactions on Parallel and Distributed Systems, 33, 805–817, 2021. a
Trott, C. R., Lebrun-Grandié, D., Arndt, D., Ciesko, J., Dang, V., Ellingwood, N., Gayatri, R., Harvey, E., Hollman, D. S., Ibanez, D., Liber, N., Madsen, J., Miles, J., Poliakoff, D., Powell, A., Rajamanickam, S., Simberg, M., Sunderland, D., Turcksin, B., and Wilke, J.: Kokkos 3: Programming Model Extensions for the Exascale Era, IEEE Transactions on Parallel and Distributed Systems, 33, 805–817, https://doi.org/10.1109/TPDS.2021.3097283, 2022. a
Tsigaridis, K. and Kanakidou, M.: Secondary organic aerosol importance in the future atmosphere, Atmospheric Environment, 41, 4682–4692, https://doi.org/10.1016/j.atmosenv.2007.03.045, 2007.
Whitby, E. R. and McMurry, P. H.: Modal aerosol dynamics modeling, Aerosol Science and Technology, 27, 673–688, 1997. a
Yarwood, G., Rao, S., Yocke, M., and Whitten, G.: Updates to the Carbon Bond Chemical Mechanism: CB05. Final Report to the US EPA, RT-0400675, https://www.camx.com/Files/CB05_Final_Report_120805.pdf (last access: 30 January 2026), 2005. a, b, c