the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 18 Jul 2023
| 18 Jul 2023
Sensitivity of tropospheric ozone to halogen chemistry in the chemistry–climate model LMDZ-INCA vNMHC
Cyril Caram
Anne Cozic
Slimane Bekki
Carlos A. Cuevas
Alfonso Saiz-Lopez
The atmospheric chemistry of halogenated species (Cl, Br, I) participates in the global chemical sink of tropospheric ozone and perturbs the oxidising capacity of the troposphere, notably by influencing the atmospheric lifetime of methane. Global chemistry–climate models are commonly used to assess the global budget of ozone and its sensitivity to emissions of its precursors, as well as to project its long-term evolution. Here, we report on the implementation of tropospheric sources and chemistry of halogens in the chemistry–climate model LMDZ-INCA (Laboratoire de Météorologie Dynamique general circulation model, LMDZ, and Interactions with Chemistry and Aerosols, INCA, version with Non-Methane HydroCarbon chemistry, vNMHC) and evaluate halogen effects on the tropospheric ozone budget. Overall, the results show that the model simulates satisfactorily the impact of halogens on the photo-oxidising system in the troposphere, in particular in the marine boundary layer. To quantify the effects of halogen chemistry in LMDZ-INCA, standard metrics representative of the behaviour of the tropospheric chemical system (Ox, HOx, NOx, CH4 and non-methane volatile organic compounds – NMVOCs) are computed with and without halogens. The addition of tropospheric halogens in the LMDZ-INCA model leads to a decrease of 22 % in the ozone burden, 8 % in OH and 33 % in NOx. Sensitivity simulations show for the first time that the inclusion of halogen chemistry makes ozone more sensitive to perturbations in CH4, NOx and NMVOCs. Consistent with other global model studies, the sensitivity of the tropospheric ozone burden to changes from pre-industrial to present-day emissions is found to be ∼20 % lower when tropospheric halogens are taken into account.
- Article
(2406 KB) - Full-text XML
-
Supplement
(822 KB) - BibTeX
- EndNote





The atmospheric chemistry of halogenated species was first studied for its importance for the composition of the stratosphere and in particular the ozone layer (Molina and Rowland, 1974; Stolarski and Cicerone, 1974), but its importance for tropospheric chemistry was also suggested as soon as the early 1980s (Chameides and Davis, 1980). With the progress of measurement techniques, marine aerosols were thought to harbour and release reactive halogens into the gas-phase (Cicerone, 1981). Later on, halogenated species were found to be responsible for ozone destruction in the Arctic troposphere (Bottenheim et al., 1990; Oltmans and Komhyr, 1986). Meanwhile, natural and anthropogenic sources of halogens were starting to be identified and characterised (Graedel and Keene, 1995; Lobert et al., 1999; McCulloch et al., 1999; Carpenter, 2003; Platt and Hönninger, 2003; Saiz-Lopez and Plane, 2004). In recent years, the addition of halogenated chemistry into global chemistry transport models has revealed its significant influence on the photo-oxidising chemistry of the troposphere on a global scale (Saiz-Lopez et al., 2012b; Simpson et al., 2015). This influence arises from the high reactivity of atomic halogens (e.g. Cl, Br, I) and halogen oxide radicals (e.g. ClO, BrO, IO and higher oxides), which are produced in the atmosphere by the decomposition of very short-lived halogenated substances (VSL) and by heterogeneous reactions on sea salt aerosols and which, in the case of iodine, are also released via ozone deposition to the ocean surface. They can also be generated by polar-specific halogen activation mechanisms on sea ice surfaces, blowing snow and the snowpack (as reviewed by Abbatt et al., 2012).
Details on the tropospheric chemistry of chlorine, bromine and iodine can be found in review articles (e.g. Saiz-Lopez and von Glasow, 2012; Simpson et al., 2015). Reactive halogen species mainly affect tropospheric chemistry by (1) modifying the partitioning of HOx (HO2+ OH) and nitrogen oxides (NOx= NO2+ NO) and reacting with NO2 to form species that remove NOx from the system via deposition or other loss processes, (2) being involved in the oxidation of non-methane volatile organic compounds (NMVOCs) and mercury, (3) participating in the formation of new particles (Giorgi and Chameides, 1985; O'Dowd et al., 2002; von Glasow et al., 2004; Gómez Martín et al., 2020; Saiz-Lopez and Von Glasow, 2012), and (4) constituting a sink for tropospheric ozone at the global scale. Halogenated species play a role in the ozone and hydroxyl radical (OH) budgets and therefore influence the oxidative capacity of the troposphere (Iglesias-Suarez et al., 2020; Badia et al., 2021). In addition, by influencing the abundances of tropospheric ozone, which is an important greenhouse gas, halogens can indirectly affect the climate (Saiz-Lopez et al., 2012a; Hossaini et al., 2015; Sherwen et al., 2017).
The loss of tropospheric ozone by halogens is mainly induced by iodine and bromine species with a lower contribution of chlorine species (Sherwen et al., 2016b). It can be caused by direct catalytic ozone destruction and reduction in ozone production through the removal of NO2 by halogen radicals. Halogen chemistry is thought to be especially active in pristine atmospheres that host 60 % to 70 % of the global burden of tropospheric ozone (Holmes et al., 2013). Based on model calculations, the halogen-driven ozone loss is estimated to be around 30 % in the marine boundary layer (von Glasow et al., 2002, 2004; Saiz-Lopez et al., 2014; Sherwen et al., 2016b) and 5 % to 20 % globally (Yang et al., 2005; Saiz-Lopez et al., 2012a, 2014; Sherwen et al., 2016b), going up to 25 % to 30 % in Badia et al. (2019). Consequently, it might be important to consider halogens in chemistry–climate modelling. It could also help to reconstruct more reliably past atmospheric compositions (Saiz-Lopez et al., 2014; Young et al., 2013).
Despite its potential importance, only three global chemistry models have so far implemented detailed representations of halogenated (Cl, Br and I) tropospheric chemistry: CAM-Chem (Saiz-Lopez et al., 2012), GEOS-Chem (Sherwen et al., 2016b) and TOMCAT (Hossaini et al., 2015). Halogen chemistry is complex with a high computational cost (Fernandez et al., 2021), Earth system models used to assess how the evolution of atmospheric composition influences climate need to find the right compromise between the level of details (e.g. number of species and processes) here for halogen chemistry representations and the computing efficiency (box 6.1 in Szopa et al., 2021). Indeed, the computational cost has to be sufficiently low for multiple, long chemistry–climate simulations. So far, none of the chemistry–climate models involved in recent large intercomparison projects included tropospheric chemistry of very short-lived halogens and hence the impacts of their chemistry on key short-lived climate forcers (such as ozone and methane). Such multi-model projects are important. They are used to assess the budget of key chemical species, which can impact directly or indirectly climate or global air quality, and their sensitivity to changes in anthropogenic and natural emissions. The results are also used to evaluate the effects of international policies (e.g. the Gothenburg Protocol to Abate Acidification, Eutrophication and Ground-level Ozone) by quantifying source–receptor relations at continental scales (in the case of the Hemispheric Transport of Air Pollution project, HTAP; Fiore et al., 2009), to explore the sensitivity of short-lived climate forcers to changes in emissions of their precursors since the pre-industrial period, or to project their possible future evolutions under a range of scenario assumptions: in the case of the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP; Young et al., 2013) or, more recently, the Aerosol Chemistry Model Intercomparison Project (AerChemMIP; Stevenson et al., 2020).
The primary purpose of this work is to assess whether iodine, bromine and chlorine chemistry can, beyond their already established effect on global tropospheric ozone budget, affect the sensitivity of ozone to the changes in its precursors. In order to do so, we implemented a simplified representation of tropospheric halogen chemistry in a global chemistry–climate model, the LMDZ-INCA model (Laboratoire de Météorologie Dynamique (LMD) general circulation model, LMDZ, and Interactions with Chemistry and Aerosols, INCA) with a focus on reproducing the overall effect of halogen chemistry on global tropospheric ozone. Results of the three global tropospheric chemistry models that already include detailed representations of this chemistry are used to guide the choices of the chemical reactions and emissions in our simple halogen scheme and to assess the LMDZ-INCA simulations. First, we evaluate the ability of the model to simulate the global tropospheric budget of halogens and their effect on ozone by using the results of the CAM-Chem and GEOS-Chem models as references. The limitations of our model are also discussed, notably how some simplifications in our model affect the simulation of the spatial distribution of some specific halogenated species. Second, we investigate how the sensitivities of ozone, methane and hydroxyl radicals to perturbations in anthropogenic emissions of photo-oxidant precursors change when halogen chemistry is accounted for. For instance, we explore changes in ozone burden and net destruction in simulations considering present-day (PD) and pre-industrial (PI) emissions with and without halogen chemistry. Finally, the last section recalls key findings, and we conclude with implications for chemistry–climate simulations of tropospheric ozone evolution.
2.1 Model description
2.1.1 LMDZ-INCA
LMDZ is an atmospheric general circulation model (GCM) developed by Sadourny and Laval (1984). The primitive equations of the GCM are solved with a time step of 3 min, a large-scale transport of tracers is carried out every 15 min, and the physical processes are calculated at a time interval of 30 min. The LMDZ model provides at each time step the physical quantities (temperature, pressure, water vapour, turbulence, boundary layer, etc.) needed to compute the transport and chemistry in INCA.
INCA is an atmospheric chemistry model developed at LSCE (Laboratoire des Sciences du Climat et de l'Environnement). The model takes into account the primary emissions from natural sources or by anthropogenic activities. It computes physical processes such as dry and wet deposition and photochemical reactions (Hauglustaine et al., 2004). The atmospheric concentration fields are integrated in time by solving differential equations when called by LMDZ, with a time step of 30 min. For this development, we used a version of INCA (version v_5.2.1) simulating interactively the gaseous photochemistry, whereas aerosol distributions obtained with a more complex version of INCA are prescribed through climatologies. This version represents in particular the chemistry of the inorganic species Ox, NOx and HOx, as well as CH4, CO, non-methane volatile organic compounds (NMVOCs) and their photo-oxidised products (Folberth et al., 2006).
Biogenic emissions like those of isoprene are provided by the global vegetation model ORCHIDEE (Organising Carbon and Hydrology In Dynamic Ecosystems) (Folberth et al., 2006). Anthropogenic emissions are based on the Community Emissions Data System (CEDS) v2016-07-2 emission inventory for the year 2010 (Hoesly et al., 2018).
The LMDZ-INCA model has been used and assessed in numerous previous model intercomparison exercises aimed at simulating the pre-industrial, present and future tropospheric composition (e.g. Szopa et al., 2013); the long range transport of air pollution in the HTAP exercise (Fiore et al., 2009); or the chemistry–climate interactions in ACCMIP (Naik et al., 2013).
2.1.2 Halogen chemistry
The chemistry of halogenated compounds implemented in LMDZ-INCA is essentially based on the scheme developed for CAM-Chem (Ordóñez et al., 2012; Prados-Roman et al., 2015a; Saiz-Lopez et al., 2012a, 2014, 2015) and GEOS-Chem (Sherwen et al., 2016a), with a priority given to the reactions of importance for the ozone sinks (Fig. 1).
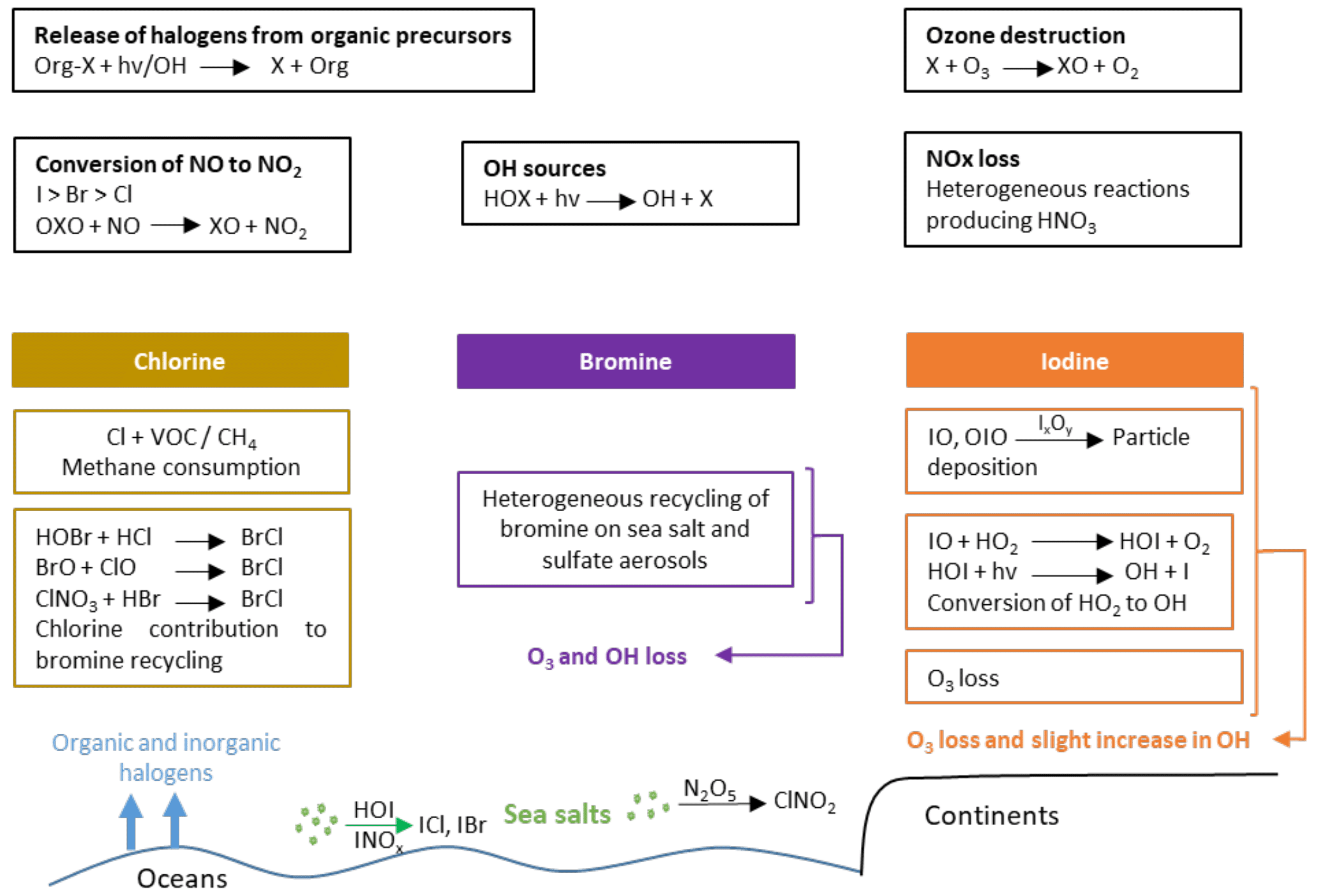
Figure 1Schematic of the most important tropospheric chemical processes involving halogenated compounds (containing Cl, Br, I) as represented in LMDZ-INCA.
A total of 42 halogenated species have been added to the INCA chemical scheme (Table 1). The list of the 160 chemical reactions added to the INCA chemical scheme can be found in Tables 2, 3, 4 and 5. Photolysis reactions of halogens as well as relative quantum yields and absorption cross-sections are listed in Table 2. Uptake coefficients for heterogeneous reactions are based on experimental values and data selected for other models and are reported in Table 5. Recycling through heterogeneous chemistry on sea salt and sulfate aerosols is included, while the heterogeneous bromine reactions in water droplets and on ice crystals in clouds are not. Dry deposition is calculated with the Wesely scheme (Wesely, 1989), while washout of gases by precipitation is simulated by using Henry's law constants detailed in Table S1 in the Supplement. Some VSL halocarbons, CH2BrCl, CHBr2Cl and CHBrCl2, represent a few percent of the emissions of halocarbons (Ordóñez et al., 2012). Considering the high uncertainty in their emissions and their limited abundances, we chose not to consider their chemistry in this version.
Table 1List of halogen tracers in INCA.
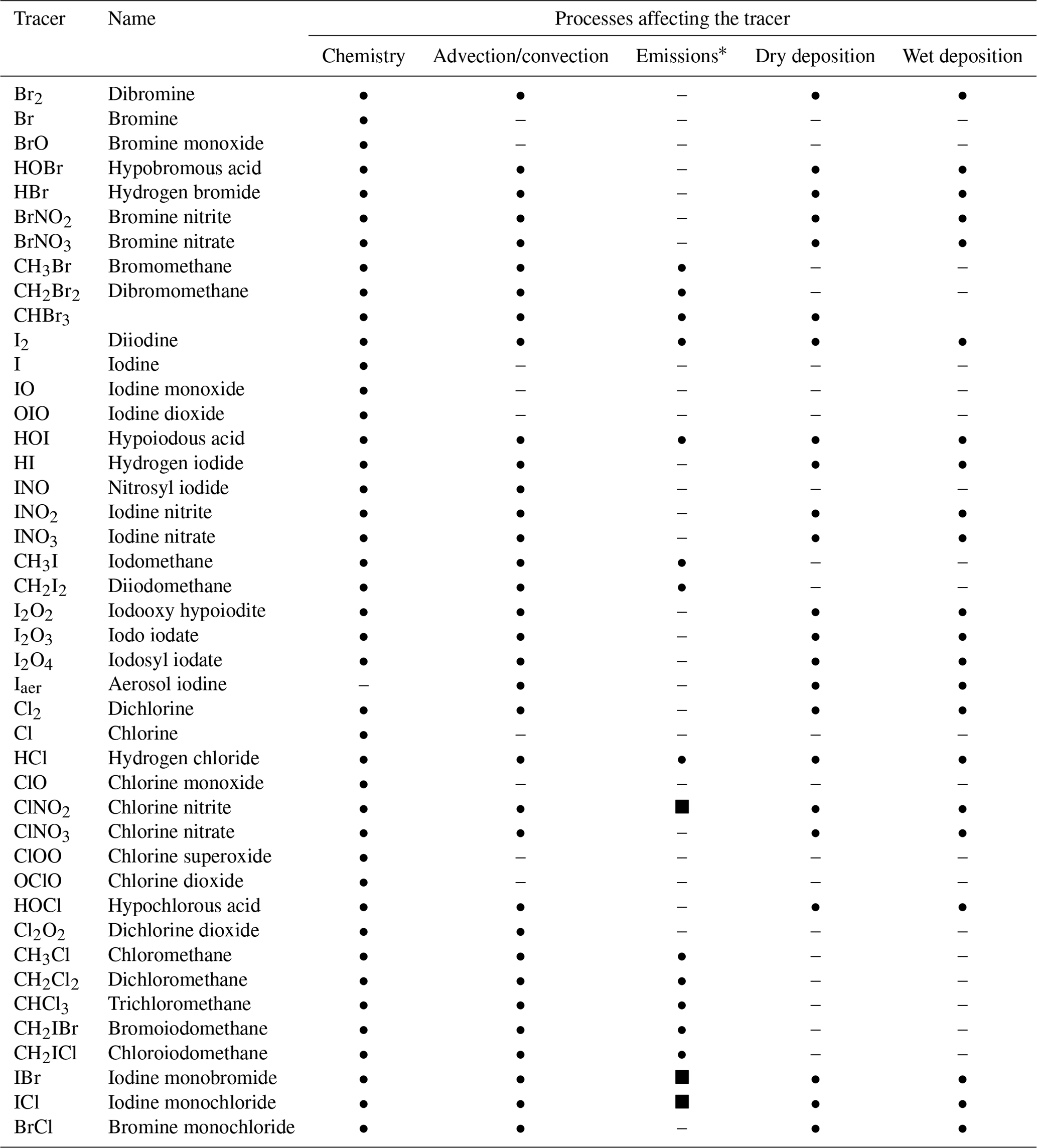
∗ Surface source • or net chemical source from sea salts (dynamically represented in INCA) ▪.
Table 2Photolysis reactions of halogens included in the scheme.

a φ1 (λ<308 nm) = 0.6, φ1 (λ=308–364 nm) = (nm) − 1.60, φ1 (λ>364 nm) = 1.0
b φ2 (λ) = 1−φ1(λ)
Table 3Bimolecular halogen reactions included in the INCA scheme. The rate constant is calculated using a standard Arrhenius expression .
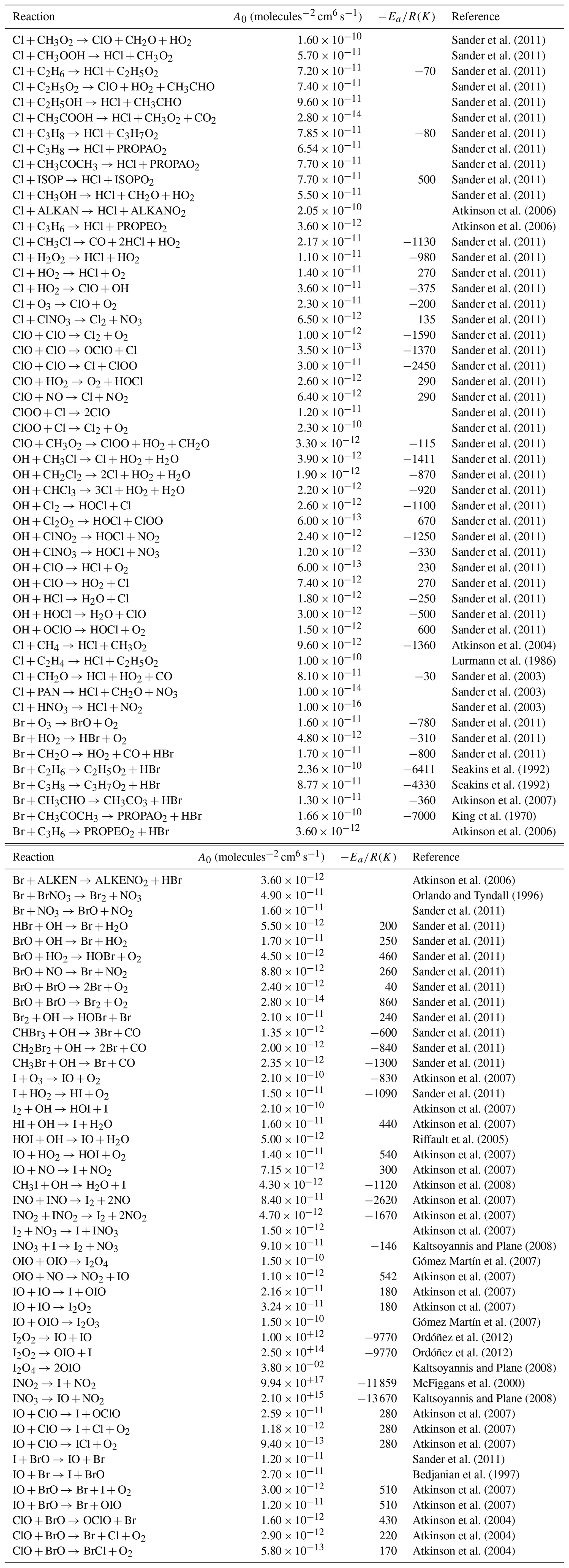
The reactions come from GEOS-Chem (Parrella et al., 2012; Eastham et al., 2014; Schmidt et al., 2016; Sherwen et al., 2016a, b) and TOMCAT (Hossaini et al., 2016) and from Thomas et al. (2011). Lumped species are detailed in Folberth et al. (2006).
Table 4Termolecular halogen reactions included in the INCA scheme.
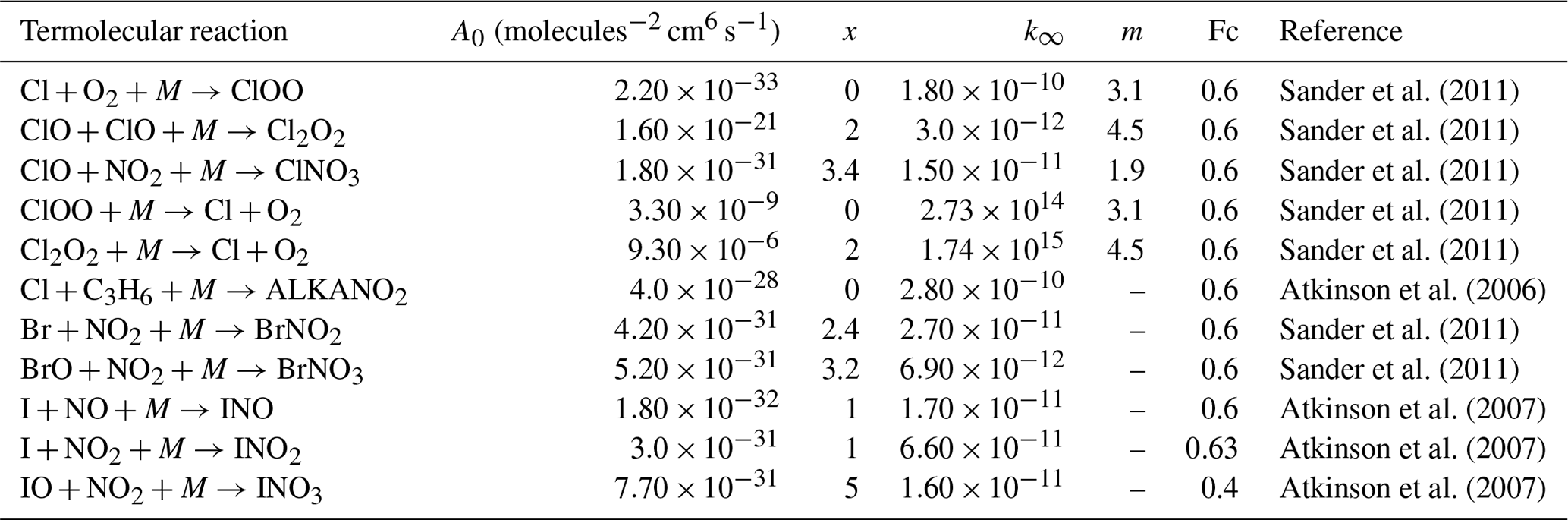
The reactions come from previous updates to halogen chemistry in GEOS-Chem (Parrella et al., 2012; Eastham et al., 2014; Schmidt et al., 2016; Sherwen et al., 2016a, b). The lower pressure limit rate (k0) is given by . The high pressure limit rate is given by k∞. Fc characterises the fall-off curve of the reaction as described by Atkinson et al. (2007).
Table 5Halogen multiphase reactions and reactive uptake coefficients (γ).
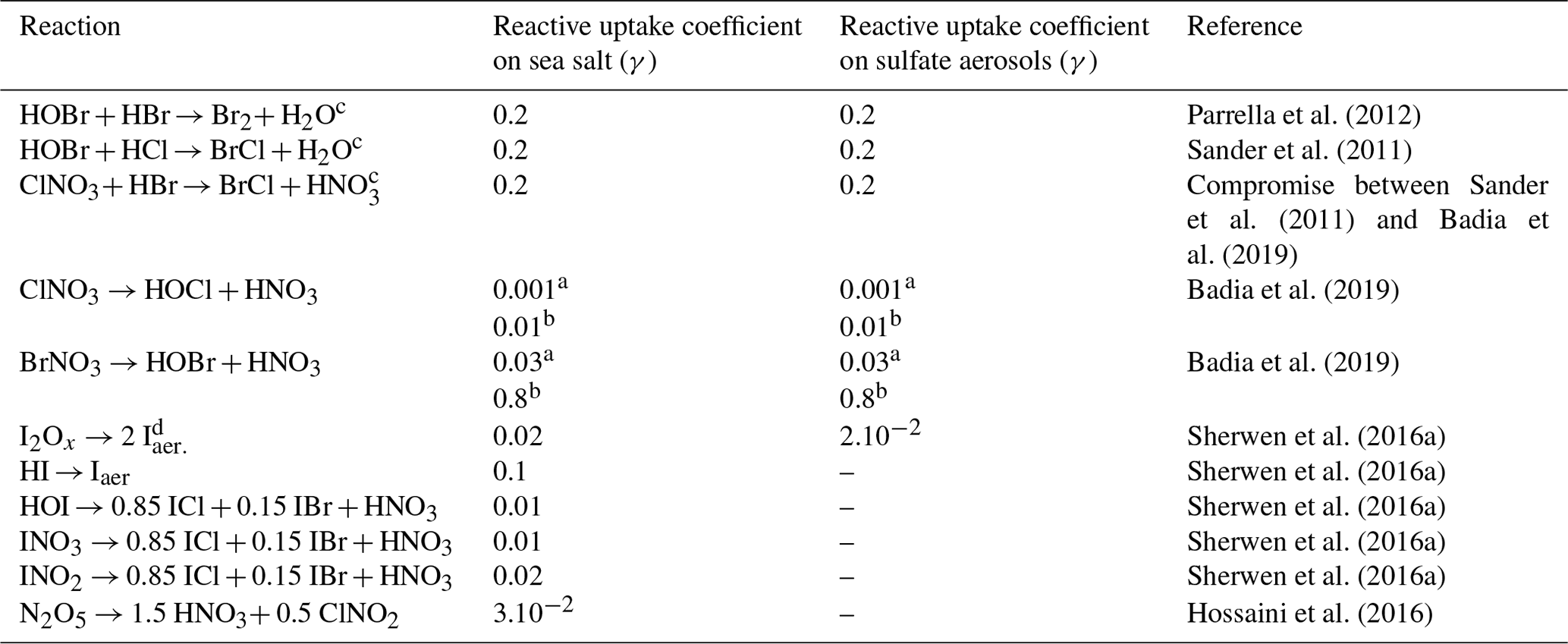
a Uptake coefficient for moderate temperatures. b Uptake coefficient for cold temperatures. c For second-order reactions, rate constants are calculated by assuming that the first reactant is limiting, and thus the first-order rate constant is divided by the concentrations of the adsorbed species. d I2Ox represents I2O2, I2O3 and I2O4.
2.2 Simulations
For these developments, LMDZ-INCA is used with a 3.75∘ longitude × 1.9∘ latitude resolution over 39 vertical layers (up to 70 km with 15 levels in the stratosphere). This resolution, coarser than the resolution now commonly used in the CMIP6 model exercise, was the standard LMDZ resolution for CMIP5 (Hourdin et al., 2013) and allows faster simulations necessary in model development. The LMDZ wind fields and sea surface temperatures are nudged on ECMWF reanalysis for the year 2010 in all simulations. The aerosol distribution and monthly variations used for heterogeneous reaction calculations are specified from climatologies precomputed with another version of the same model but with fully interactive calculations of aerosols. In this version, the stratospheric composition is prescribed. All simulations cover 1 year (2010) and follow a spin-up period of 1 year. Three types of simulations have been performed to simulate (i) present-day conditions, (ii) pre-industrial conditions and (iii) sensitivity to ozone precursors. The surface emissions of NOx, CO and NMVOCs and CH4 concentrations considered in the present-day and pre-industrial simulations are presented in Table S2. For sensitivity tests, simulations with individual emission perturbations of photo-oxidant precursors (−20 %) of NOx, CO and NMVOCs and concentration reduction (−20 %) of CH4 were performed. For each type of simulation, two versions of the model are applied: one accounting for the halogen chemistry (called Halo simulations hereafter) and a reference simulation without halogen chemistry called “NoHalo”.
2.2.1 Present-day set-up
Primary halogenated compounds can originate from both inorganic and organic sources. Inorganic species are reactive species emitted by different sources (e.g. oceans, sea salts) but also result from atmospheric reactions (Simpson et al., 2015). Organic species are only of primary origin and are photo-oxidised in the atmosphere to form halogen radicals (Simpson et al., 2015). Table 6 shows the emissions of halogenated compounds as considered in INCA. Organo-bromines (e.g. CHBr3, CH2Br2), organo-iodines (e.g. CH3I, CH2I2) and inter-halogen species (e.g. CH2IBr, CH2ICl) are those used by the CAM-Chem model (Ordóñez et al., 2012).
Sources of organic chlorine are diverse, mainly originating from biological activity on the surface of the oceans and from forest fires. The emissions of these chlorocarbons are detailed in Table S2. In INCA, inorganic chlorine, HCl – the major reservoir of chlorine in the atmosphere – is considered to be emitted by sea salts (90 000 Gg Cl yr−1) and by forest fires (6400 Gg Cl yr−1) with total based on Lobert et al. (1999) and Hossaini et al. (2016). Note that more comprehensive but hard to implement approaches relying on pH-dependent parameterisation of the dechlorination of sea salts have been recently implemented in GEOS-Chem. HCl emissions from industrial sources (McCulloch et al., 1999) are neglected as recommended by Wang et al. (2019) to avoid overestimating HCl in urban areas. ClNO2 is emitted by the heterogeneous reaction of N2O5 on sea salts and produces 2217 Gg Cl yr−1.
CH3Br emissions are from oceanic origin (40 %) and from agriculture (60 %) (Barker et al., 2016) and represent 91 Gg of Br per year (Schmidt et al., 2016). IBr and ICl originate from the heterogeneous reaction of HOI, INO2 and INO3 on sea salt aerosols (Table 5) as considered in other models (Saiz-Lopez et al., 2014; Sherwen et al., 2016a) but without accounting for pH dependence (McFiggans et al., 2000).
The inorganic iodine distribution of emissions (HOI and I2) are prescribed from a climatology computed by the CAM-Chem model considering sea–air fluxes as a function of the oxidation of aqueous iodide by atmospheric ozone on the ocean surface parameterised by Carpenter et al. (2013) and MacDonald et al. (2014) and which depends on the ozone deposition to the ocean surface, the sea surface temperature and the wind speed (Prados-Roman et al., 2015b).
2.2.2 Pre-industrial set-up
The aim of this simulation is to explore how the sensitivity of ozone to anthropogenic emissions perturbations have changed since the pre-industrial period (a present-day climate is thus considered). Only emissions from natural sources are included for the simulation of the pre-industrial troposphere, following the approach used by other modelling teams to quantify this perturbation. Emissions of halogenated compounds from biomass burning are reduced to 10 % of their current values (Wang and Jacob, 1998). The methane concentration is set at 700 ppbv (IPCC, 2013). Emissions of biogenic volatile organic compounds are kept at their present-day level due to uncertainties in the effect of their drivers over the past century and the resulting high uncertainty regarding the quantification of change in their emissions (Szopa et al., 2021).
Organo-iodine emissions are almost unchanged between the PI and PD. For organo-bromines, pre-industrial emissions of CHBr3 and CH2Br2 are equivalent to the PD ones, following the methodology of Parrella et al. (2012). The concentration of CH3Br is set to ∼5 ppbv and is considered to be in agreement with the measurements from ice cores (Saltzman et al., 2004). Pre-industrial emissions of CH3Cl, CHCl3 and CH2Cl2 are from Carpenter et al. (2014), Hu (2012), Montzka et al. (2010), and Worton et al. (2006). HOI and I2 emissions, pre-computed by CAM-Chem, are ∼40 % lower than today because they are sensitive to atmospheric ozone content which was lower in the pre-industrial era (Prados-Roman et al., 2015a; Sherwen et al., 2017). Global emissions for each primary halogenated compound are reported in Table 6.
Table 6Emission of halogen gases for pre-industrial and present-day simulations.
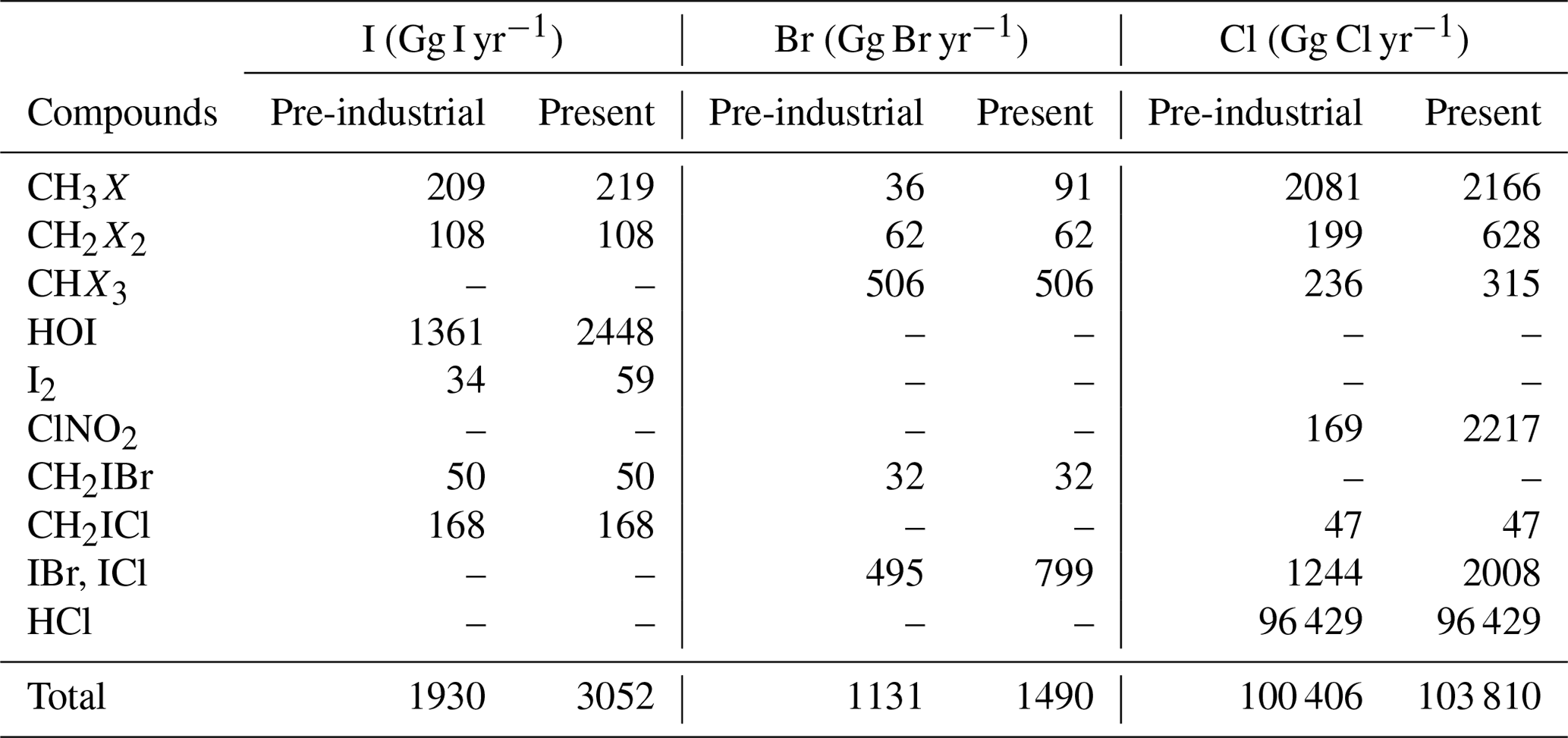
Sources of organic species (CH3X, CH2X2, CHX3) are shown in terms of emissions. I2 and HOI are the inorganic oceanic source that depends on surface ozone (Carpenter et al., 2013). IBr and ICl are released following gaseous iodine uptake on sea salt. ClNO2 is emitted after absorption of N2O5 on sea salt.
We first assess the ozone sensitivity to halogen chemistry in LMDZ-INCA, and then we evaluate the performance of the model in simulating the climatology of halogen species which are important for the global ozone loss. In order to do so, model outputs for compounds of interest in this chemistry are compared to a range of halogen observations and simulations from other models with halogen chemistry. An analysis of the model tropospheric budgets of key halogen species is also performed.
3.1 Impact on ozone loss in the present day
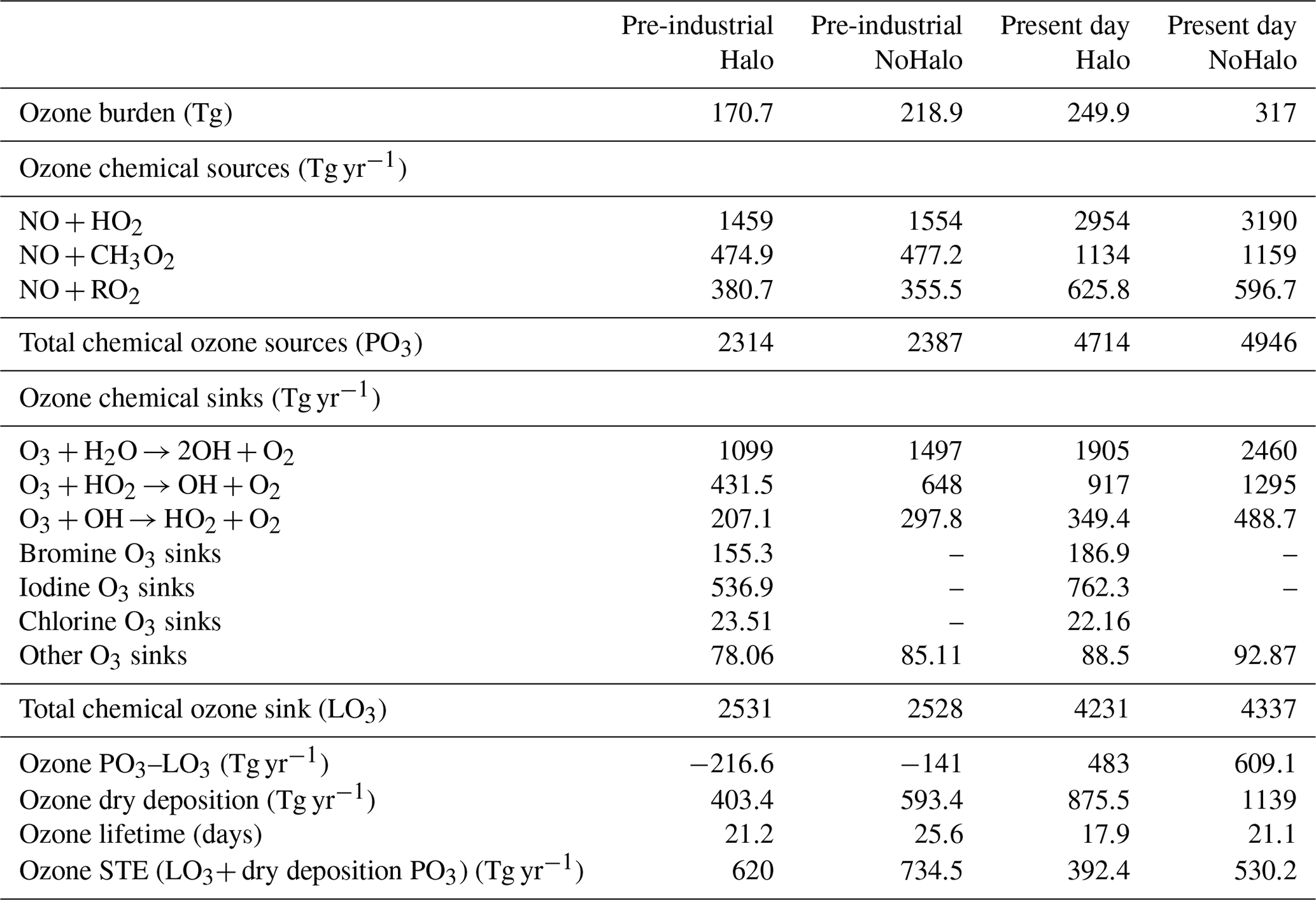
Table 7 presents the ozone budget in LMDZ-INCA with and without consideration of halogen chemistry. Overall, when halogens are considered, the tropospheric ozone burden is reduced by 22 % for present-day conditions, which is in the 5 % to 25 % range reported from other models which include Cl, Br and I chemistry (Saiz-Lopez et al., 2012a, b, 2015; Sherwen et al., 2016a; Badia et al., 2019, 2021). Over the global troposphere, it is associated with a decrease in the annual-average ozone mixing ratio (mass-weighted) of ozone of 9.7 ppbv (compared to 9.4 ppbv in GEOS-Chem; Sherwen et al., 2016a). Over the tropospheric tropical column, the annual-average ozone mixing ratio (mass-weighted) decreases by 8 ppbv (21 %), which is twice as large as the relative change reported in Saiz-Lopez et al. (2014). Surface ozone averaged over continental areas undergoes a decrease of 6 ppbv (23 %), while surface ozone in oceanic areas, where the majority of primary halogenated compounds are emitted, decreases by 7.7 ppbv (32 %) (range reported in other studies is 20 %–33 %; Saiz-Lopez et al., 2014; Long et al., 2014; Prados-Roman et al., 2015a; Sherwen et al., 2016a).
Figure 2 shows the absolute changes in surface ozone and relative change in zonally averaged ozone in a simulation with halogen chemistry relative to a simulation without it. Significant relative losses are found in the Southern Hemisphere, which are due to the strong emissions of halogenated species from the ocean in this region (Long et al., 2014; Saiz-Lopez et al., 2012a; Schmidt et al., 2016; Sherwen et al., 2016a, b) and the smaller absolute abundance of ozone. Most of the change in total tropospheric ozone concentration occurs in the free troposphere between 350 and 900 hPa (63 %). This result is similar in magnitude to the relative change reports in GEOS-Chem (65 %; Sherwen et al., 2016a) and CAM-Chem (65 %; Iglesias-Suarez et al., 2020).
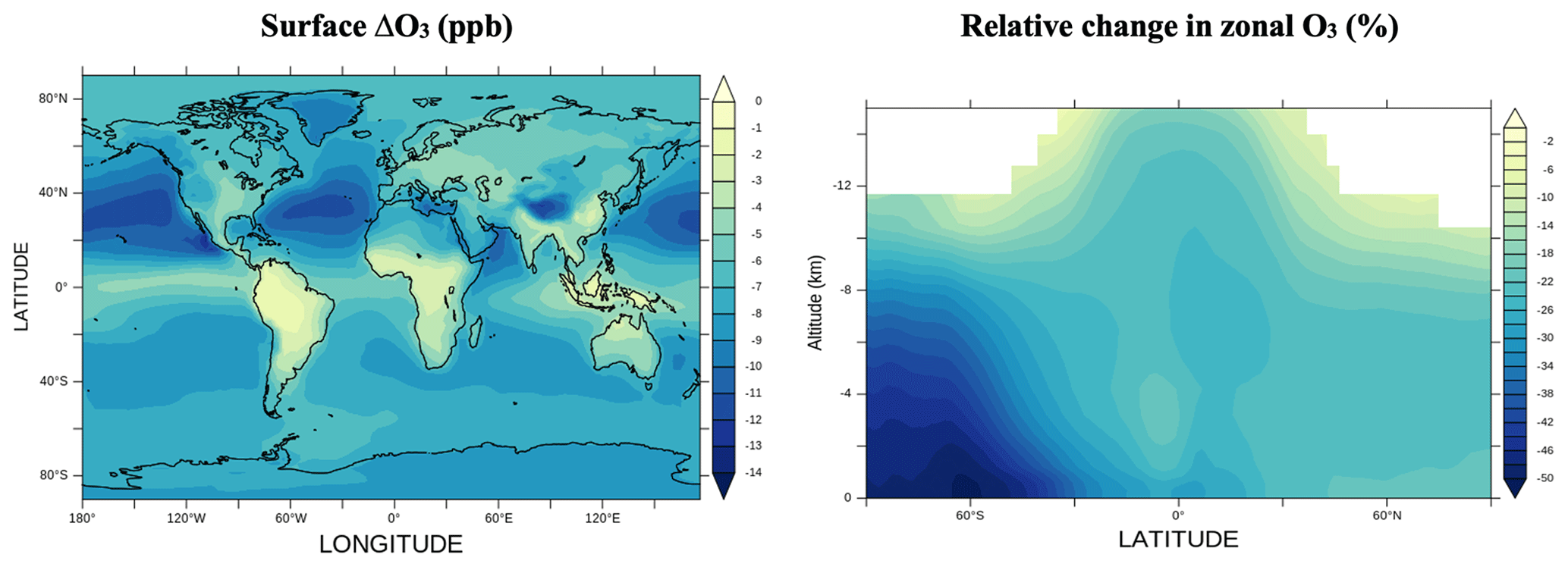
Figure 2Surface ozone changes (ppbv) and relative change in zonal tropospheric ozone (%) between simulations with and without halogen chemistry.
The annual production of ozone decreases by 4.7 % when the chemistry of halogens is considered. This decrease is due to a reduction in NOx concentrations of 32.8 % (Table 7) due to the hydrolysis of XNO3 (X = Cl, Br, I) leading to deposition losses, in accordance with previous studies (Saiz-Lopez et al., 2012a; Long et al., 2014; von Glasow et al., 2004; Parrella et al., 2012; Schmidt et al., 2016; Sherwen et al., 2016a).
The vertical profile of the different contributions to chemical ozone loss is shown in Fig. 3. The processes leading to ozone loss differ considerably between simulations with and without halogenated compounds (Table 7). Halogens account for nearly ∼22 % of the ozone loss in the boundary layer in the halogen simulation, as shown in Fig. 3. Halogens represent 55 % of the Ox (Ox = odd oxygen) loss in the upper troposphere (350 hPa > pressure > tropopause), while this value is about 33 % and 40 % in GEOS-Chem with the iodine plus bromine and the iodine plus bromine plus chlorine chemistry, respectively. Such differences could be partly due to differences in convection schemes which induce different uplifts of IO in the upper troposphere resulting in different IO concentrations (see Sect. 3.1.3 below) but also to different representations of the stratospheric composition or chemistry between models.
When chemistry of halogenated compounds is considered, the total annual chemical ozone loss decreases by 2.4 % (Table 7). The relative contributions of different halogens to ozone loss are very similar to those of GEOS-Chem in Sherwen et al. (2016b). The loss of ozone by chlorine, bromine and iodine represents 0.5 %, 4.7 % and 18 % of the total losses, respectively; thus, halogens represent 23.2 % of the total loss of ozone. The sum of the ozone loss caused by the halogens is 984 Tg yr−1. Iodine is responsible for 78 % of that halogen-mediated loss, while bromine accounts for 19 %, which is close to the values reported by Iglesias-Suarez et al. (2020) (80 %, 16 %, respectively). Iodine is responsible for an overall tropospheric ozone loss of 762 Tg yr−1 (comparable to the 748 Tg yr−1 reported by Sherwen et al., 2016b). Much of this loss arises from the photolysis of HOI after its production from the reaction of IO with HO2 and the photolysis of OIO after its production from reactions implicating IO and IxOy. Bromine is responsible for an overall tropospheric loss of 187 Tg yr−1 which mostly arises from the photolysis of HOBr after its production from the reaction of BrO with HO2.
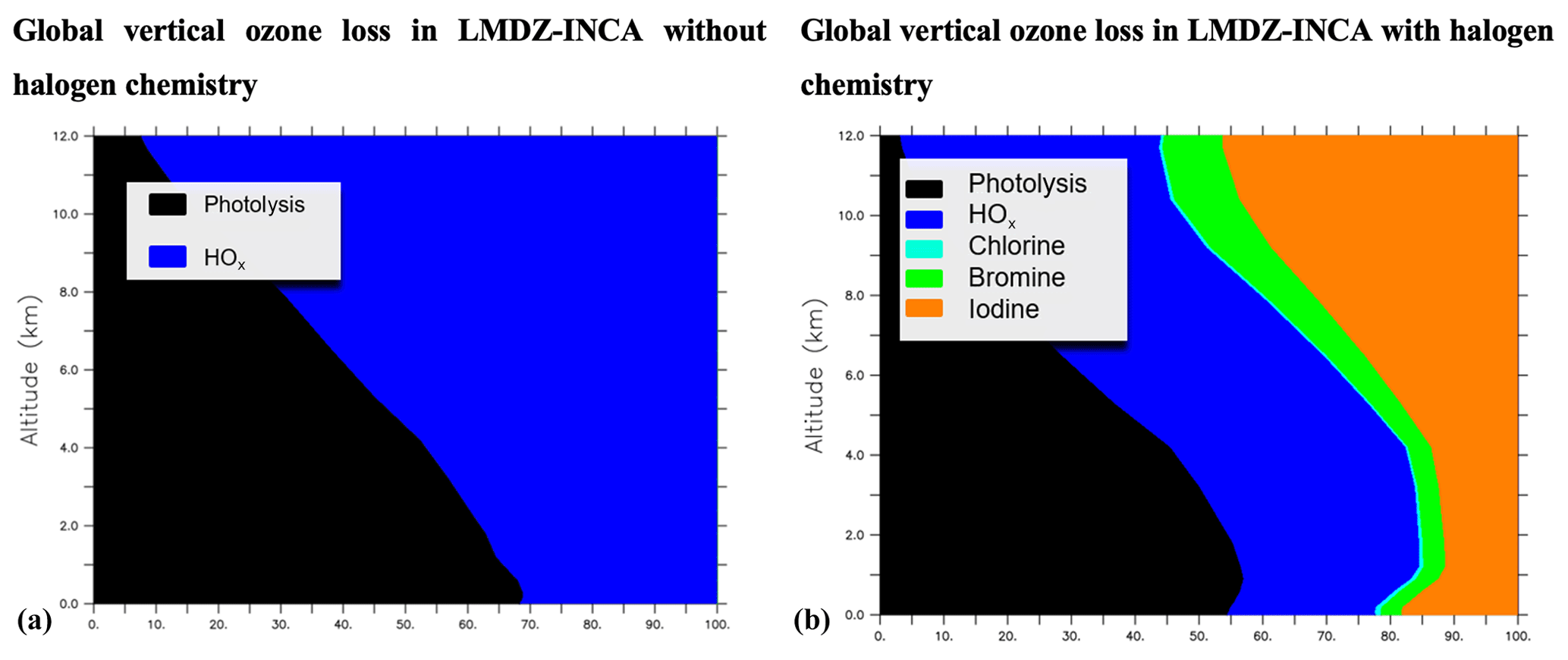
Figure 3Contribution of the chemical ozone sinks as a function of the altitude without (a) and with (b) consideration of the chemistry of halogens in LMDZ-INCA.
The inclusion of halogens leads to an overall decrease in the net production of tropospheric ozone (PO3–LO3) of 20 % (126 Tg yr−1). Consequently, the lifetime of ozone (= tropospheric burden losses) is significantly decreased by 3.2 d and reaches 17.9 d.
Table 7Comparison of global tropospheric Ox budgets of “Halo” and “NoHalo” simulations in INCA for the PI and PD. STE signifies stratosphere–troposphere exchange.
3.2 Present-day budgets for halogenated species in LMDZ-INCA
In order to reflect the role played by the halogenated species in their respective cycle, inorganic halogens are presented hereafter under the name Xy which can be divided into HX (the acidic forms of halogenated compounds) and X* (composed of the nocturnal reservoirs of reactive halogenated species and the reactive halogenated species). HX in LMDZ-INCA takes part in heterogeneous bimolecular reactions. In CAM-Chem and GEOS-Chem, the representation of this chemistry is more detailed: HX can be incorporated in droplets or particles and dissociate (Ordóñez et al., 2012; Prados-Roman et al., 2015a; Saiz-Lopez et al., 2012a, 2014, 2015; Sherwen et al., 2016a). X* is subdivided into the “reactive halogen” XOx; grouping X + XO*; and the reservoir species X2, HOX, XNO2 and XNO3, which have a relatively longer lifetime. These last species are formed mostly at night and serve as reservoirs of reactive halogens far from their primary sources.
In the following subsections, we present, for each halogen, the species considered in each reservoir, their respective tropospheric mass burden and the main fluxes between these reservoirs. The purpose is to evaluate the representation of halogenated species of interest for ozone and to discuss the limitations of the model in the computation of the budget of each halogen family.
3.2.1 Iodine
Iodine chemistry is the most impactful with regard to the ozone loss. The simplified iodine cycle is shown schematically in Fig. 4. The burden of inorganic iodine excluding IBr or ICl and iodine in aerosols is ∼22.0 Gg in the troposphere in LMDZ-INCA, comparable to the value of ∼27.9 Gg reported by Sherwen et al. (2016a, Fig. 7). HOI and OIO species, whose photolysis result in atomic iodine production, are the main intermediates for direct ozone loss. HOI is the most abundant iodine species, and its wet deposition controls the total atmospheric iodine burden. Its tropospheric burden of 12.8 Gg I in INCA is very close to the 13 Gg simulated by Sherwen et al. (2016b). The tropospheric burden of OIO in INCA, 0.43 Gg I, is comparable to the 0.55 Gg I also reported in Sherwen et al. (2016b). Overall, the global levels of HOI and OIO simulated by INCA are similar to their simulation by other models described in the literature.
The tropospheric burdens of HI, INO2 and I2 sum up to 1 Gg I, lower than the burdens reported for GEOS-Chem and totalling 2.48 Gg I (Sherwen et al., 2016b). The global burden of I2 in INCA is lower than that in GEOS-Chem (Sherwen et al., 2016b) because in Sherwen et al. (2016b) heterogeneous reactions transform INO3, INO2 and HOI into I2, whereas in INCA, these reactions produce IBr and ICl, similarly to the heterogeneous reactions adopted by Sherwen et al. (2016a). Other quantifications in the literature are lacking to explain the differences in INO2 and HI; however, Saiz-Lopez et al. (2014) indicate that minor iodine species (Imin= HI + OIO + INO2+ INO) represent less than 5 % of the mass in tropospheric inorganic iodine (2.3 % in our case). In INCA, the tropospheric Iy mean is 1.3 pptv (parts per trillion volume), whereas previous model studies found a range of ∼0.5 to 1 pptv (Saiz-Lopez et al., 2014; Sherwen et al., 2016a, b). A large set of observations has been assembled by Sherwen et al. (2016b) for IO. Measurements of IO during oceanic campaigns show a strong diurnal variation (IO being present only during daytime) with climatological concentrations (local observations averaged over 20∘ bins) comprised between 0.3 and 1 pptv. These IO concentrations show a weak latitudinal variability and a rapid decrease with altitude to reach values of about 0.2 pptv from 2 km to the tropopause (observations gathered and reported by Sherwen et al., 2016b). Surface values simulated by INCA agree within a factor of 2 with these observations. Over oceans, the annual mean surface concentrations of IO is approximately 0.2 pptv with concentrations of between 0 and 2 pptv. This is similar to the range simulated by Sherwen et al. (2016b) (0.25–1 pptv) and to the observations reported by Prados-Roman et al. (2015b). In the tropics (20∘ S–20∘ N), levels of surface IO in INCA (0.35 pptv) are in agreement with model results reported for CAM-Chem (∼0.4 pptv) (Saiz-Lopez et al., 2014). In the free troposphere, IO levels slightly increase with altitude to reach diurnal values of ∼0.5 pptv at 10 km in INCA. This overestimation of IO in the upper troposphere is common to all current model-simulated iodine and remains unclear (Sherwen et al., 2016b; Badia et al., 2019).
The IOx family influences the oxidative capacity of the troposphere through the catalytic cycles of ozone depletion (Brasseur, and Solomon, 2005), modifies the HOx (HO2/OH) and NOx (NO2/NO) ratios (Bloss et al., 2010), produces photolabile higher-order iodine oxides (IxOy) (Gómez Martín et al., 2020; Lewis et al., 2020), and generates different forms of inorganic iodine (Saiz-Lopez et al., 2012b). For that reason, a correct representation of the branching ratios and the generation and loss of IOx is crucial. Globally, IOx production in INCA is dominated by inorganic photolysis of HOI (80.5 %), OIO (10.2 %), INO3 (5.4 %) and I2Ox (3.9 %). This partition is comparable to the one reported in Sherwen et al. (2016a), who found a 76 % HOI photolysis contribution and a 11 % OIO photolysis contribution. The main IOx loss pathway is the production of HOI by IO reaction with HO2 (78.5 %), with additional loss pathways by self-reaction and reactions with NOx and BrO contributing 11.3 %, 5.7 % and 4.5 %, respectively – numbers that are very close to the branching ratios reported in Sherwen et al. (2016a) (77 %, 10 %, 7.7 % and 4.6 %, respectively).
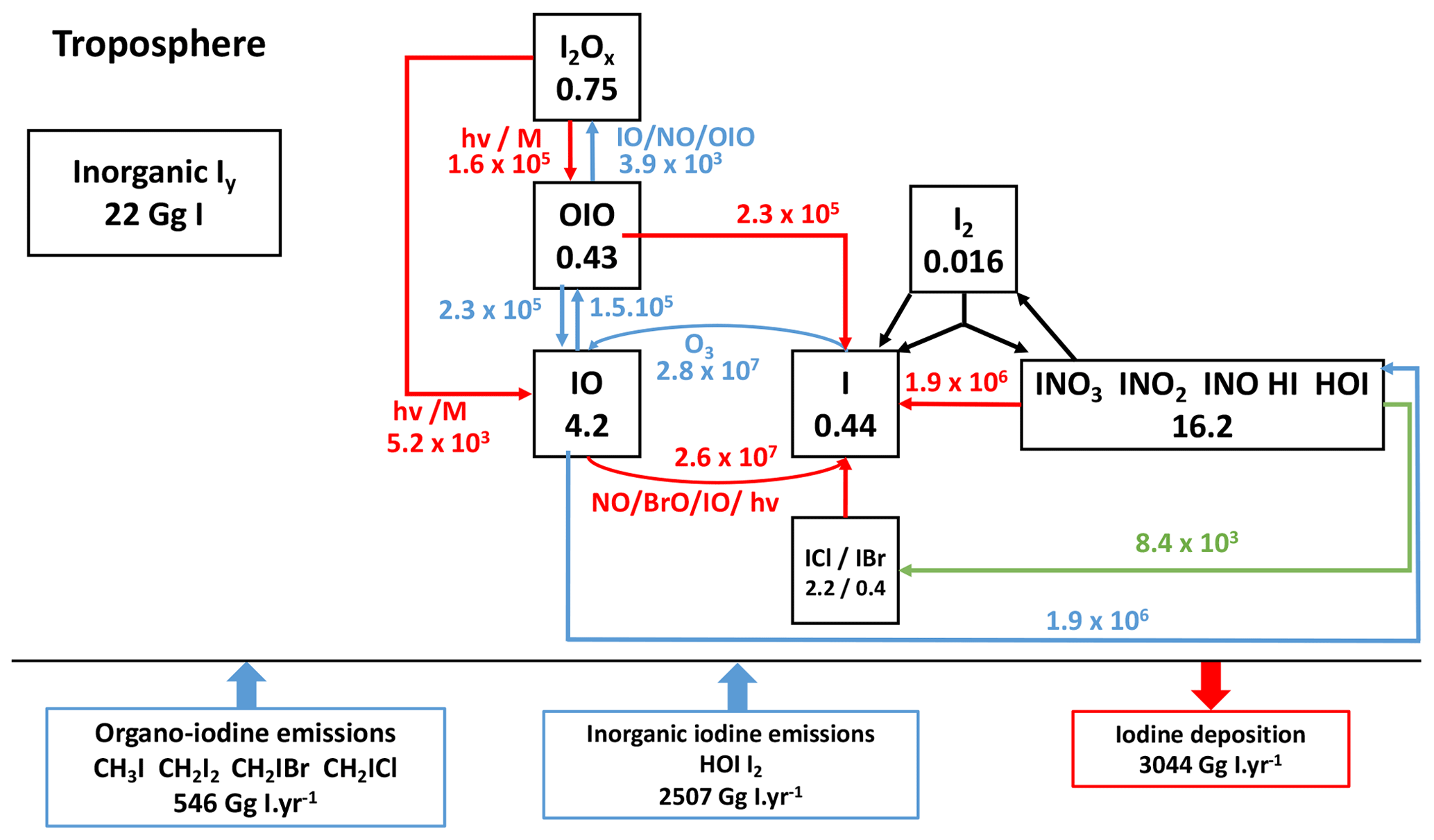
Figure 4Iodine budget in the present-day reference simulation with simplified cycle of tropospheric Iy species. Heterogeneous reactions are shown in green. Photolysis reactions are in red. The numbers in the boxes represent the mass balance of the species families (in Gg I), and the numbers near the arrows represent the fluxes through chemical reactions (in Gg I yr−1).
3.2.2 Bromine
The tropospheric bromine budget and simplified recycling cycle are illustrated in Fig. 5. The tropospheric inorganic bromine burden is ∼27 Gg in LMDZ-INCA, close to the 28 Gg reported for the most recent version of GEOS-Chem (Wang et al., 2021). On an annual average, INCA simulates daytime BrO values in the free troposphere of ∼0.3 pptv within the tropics, close to the results in CAM-Chem (∼0.2 pptv) (Fernandez et al., 2014). At the tropical tropopause layer, the modelled tropical BrO concentrations in INCA are ∼3 pptv, close to the observed concentrations of 2.0±1.5 pptv and of 3.2±1.6 pptv at two different altitudes performed by Dorf et al. (2008) and reported by Fernandez et al. (2014) for the evaluation of CAM-Chem. Overall, the global tropospheric mean of BrO simulated by INCA is 0.6 pptv (in agreement with model results from Parrella et al., 2012) but is in the lower end of the 0.5 to 2 pptv range inferred from observations (Dorf et al., 2008; Saiz-Lopez and von Glasow, 2012; Stone et al., 2012).
In this first implementation of halogen chemistry in a version of LMDZ-INCA, the focus is on gaseous chemistry. Aqueous chemistry in droplets or on ice crystals in clouds is not accounted for in the first version of the scheme because it is complex and uncertain and, more importantly, might not be a dominant mechanism for global ozone loss. Nonetheless, the scheme includes recycling through heterogeneous chemistry on sea salt and sulfate aerosols. A study by Badia et al. (2019) using WRF-Chem shows that heterogeneous halogen chemistry (on all types of aerosols) allows a better vertical Bry partitioning but contributes little (∼6 %) to ozone loss compared to the gas-phase halogen chemistry (25 %) on a global scale. Our choice to include, in this first step, reactions on sea salt and sulfate aerosols only is suited to reproduce bromine-mediated ozone loss in the marine boundary layer but probably tends to slightly underestimate the halogen-mediated ozone loss in the global troposphere.
Due to this lack of in-cloud halogen chemistry in cloud droplets and ice crystals, whose importance increases with altitude, the bromine chemistry in INCA is less dynamic and the recycling of bromine is less efficient in LMDZ-INCA compared to those reported for some versions of GEOS-Chem (Parrella et al., 2012; Schmidt et al., 2016; Sherwen et al., 2016a; Zhu et al., 2019). Note, however, that the recently updated HOBr heterogeneous chemistry implemented in the most recent GEOS-Chem version (Wang et al., 2021) results in much slower recycling of HOBr in clouds and aqueous aerosols than previously calculated. This results in fluxes from heterogeneous reactions (specifically HOBr + HBr → Br2+ H2O and HOBr + HCl → BrCl + H2O) that sum up to 32 500 Gg Br yr−1, which is smaller for example than our estimates of 86 000 Gg Br yr−1 but remains of the same order of magnitude. Finally, our branching ratios are found to be consistent with those in Schmidt et al. (2016) and Wang et al. (2021). This leads us to conclude that, although LMDZ-INCA does not integrate the same degree of details in tropospheric halogen heterogeneous chemistry compared to its peers, global fluxes in the bromine cycle of INCA appear to be more comparable in terms of the bromine cycle with those in the newest version of GEOS-Chem (see the detailed bromine budget and cycle in Fig. S1) and CAM-Chem (Iglesias-Suarez et al., 2020) than fluxes in the older versions of GEOS-Chem (Parrella et al., 2012; Schmidt et al., 2016; Sherwen et al., 2016a; Zhu et al., 2019). All these estimates remain of the same order of magnitude, but the range of fluxes illustrates the level of uncertainties about the strength of the halogen heterogeneous recycling.
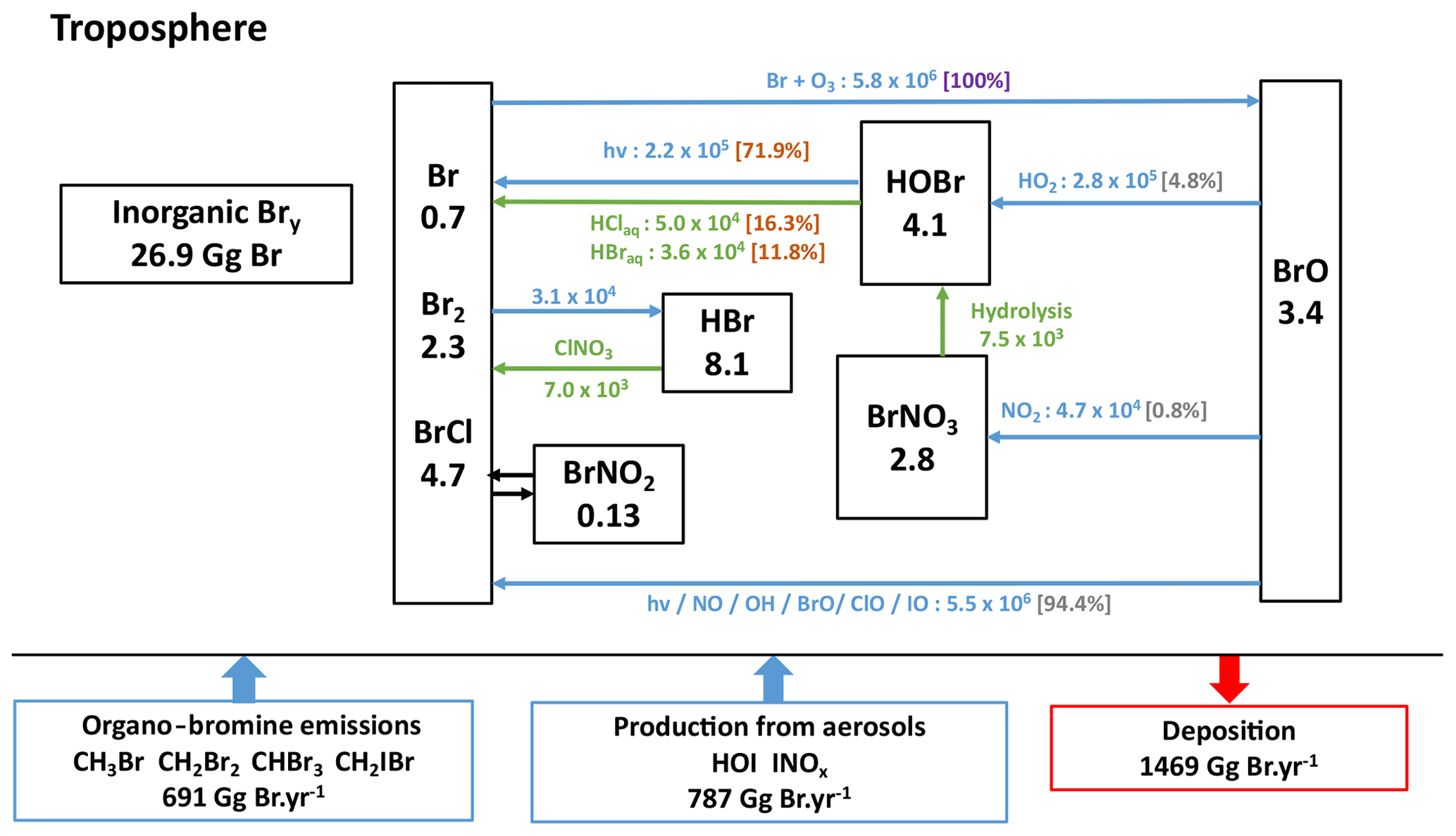
Figure 5Bromine budget in the present-day reference simulation with a simplified representation of the cycle of tropospheric Bry species. Fluxes through the reactions, indicated next to the arrows, are in gigagrams Br per year (Gg Br yr−1). Heterogeneous reactions are represented in green. Percentages in brackets represent the branching ratios for the Br chemical sink (in violet), for the BrO chemical sinks (in grey) and for HOBr chemical sinks (in light maroon). The numbers in the boxes represent the burden of the species or families in gigagrams Br (Gg Br), and the numbers near the arrows represent the fluxes through chemical reactions in gigagrams Br per year.
3.2.3 Chlorine
The total tropospheric burden of Cly (inorganic chlorine) is 290 Gg of Cl, largely dominated by HCl. The total load of HCl is smaller than the one reported for GEOS-Chem (Wang et al., 2019) essentially because of the difference in the representation of the emissions. However, the total burden of reservoir species has a similar magnitude (10.4 Gg Cl). The halogen chemistry in INCA leads to a 25 % higher quantity of available reactive chlorine compounds: Cl and ClO* (ClO* being ClO + ClOO + OClO + 2Cl2O2).
Atomic chlorine has two main chemical sinks. The first and most important in terms of reaction flux is the transformation of atomic Cl into ClO*. This conversion takes place mainly by reaction with ozone (99.5 %), while the conversion of ClOx to Cl is dominated by reaction with NO (93.9 %). The second path is represented by the chemical loss of reactive chlorine Cl to inactive chlorine HCl (Fig. 6), which is due to the reactions of Cl with methane and several NMVOCs. The conversion of Cl to ClO* is in strong competition with the second route. By dividing the fluxes of the conversion Cl to ClO* by the flux of conversion of Cl to HCl, we find that the chain length is short and equal to 1.3 (4.8×104/3.6×104). This is close to the chain length of 1.6 calculated by Wang et al. (2019) and shows that the branching ratios are of the same order of magnitude in both models despite differences in NMVOC and CH4 concentrations between the two models. Overall, we find that the partition between Cl and ClO* is correct in the model as well as the balance between radical and non-radical species, although the latter is a little more shifted towards reactive species in INCA compared to GEOS-Chem and CAM-Chem.
The average of climatological observations (mean over several months and representative of large areas) over six oceanic and coastal remote locations shows a mean surface HCl mixing ratio of 196±114 pptv, whereas INCA simulates an average of 193±98 pptv, compared to the 246±90 pptv in GEOS-Chem (Wang et al., 2019) (see Fig. S2). However, for continental regions, LMDZ-INCA underestimates the HCl concentrations compared to observations mainly because it does not include industrial emissions. ClNO2 over continental areas does not exceed a few tens of pptvs in INCA, whereas observations at several continental locations, as gathered by Wang et al. (2019) (their Table 5), show concentrations of a few hundreds to thousands of pptvs over such locations for hourly maxima. This difference can arise from the coarse resolution of the model which does not resolve the spatial heterogeneity of these local observations (Sherwen et al., 2016a) and the absence of ClNO2 production in sulfate–nitrate–ammonium aerosols (Wang et al., 2019). The chlorine Cl* measurements to which we compare LMDZ-INCA results represent the sum of Cl2, HOCl, ClNO2, ClNO3 and other minor components as explained in Pszenny et al. (1993). LMDZ-INCA Cl* values are of the same order of magnitude as measurements (see Table S3).
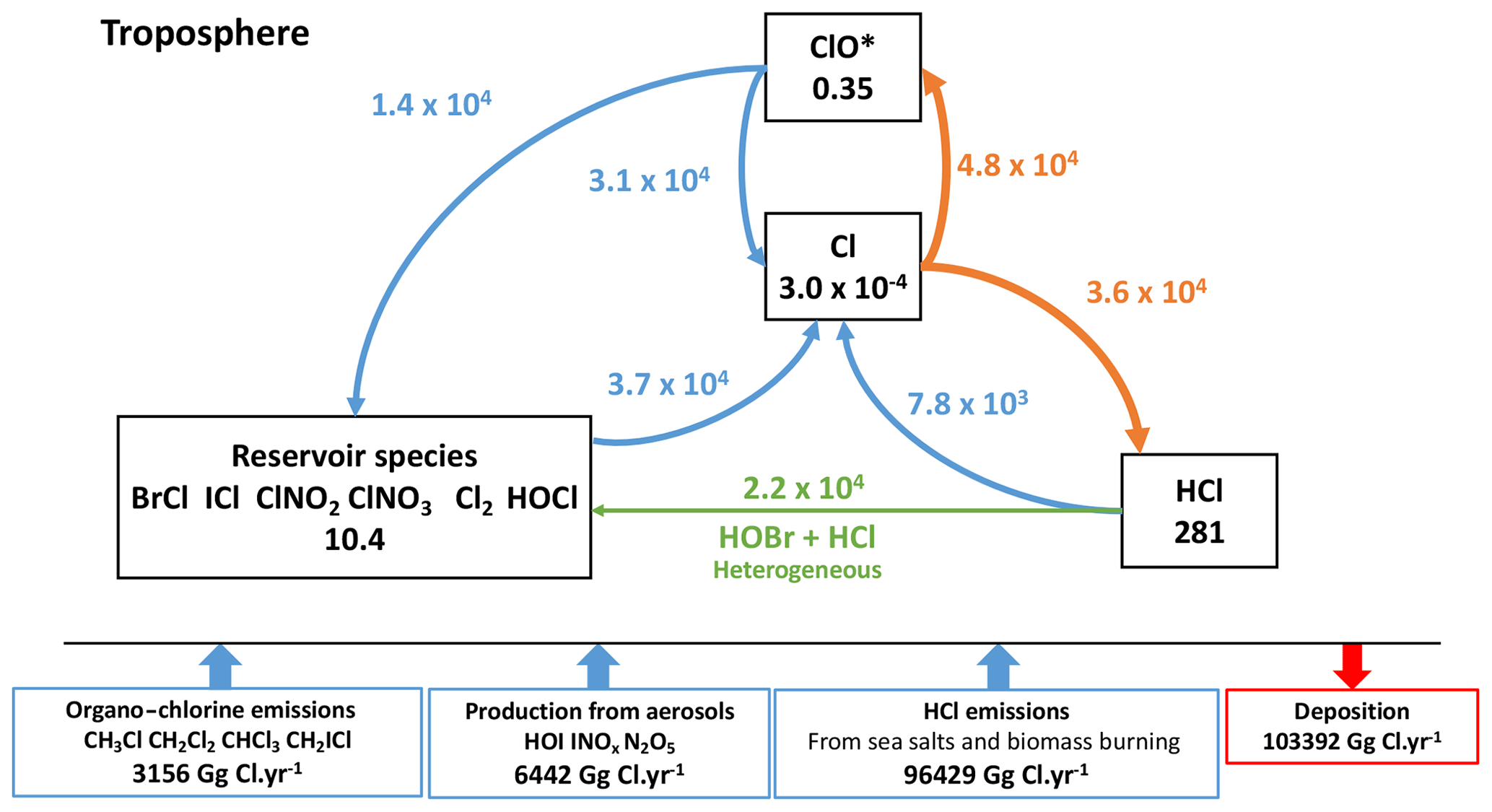
Figure 6Simplified representation of the cycle of tropospheric Cly species in the reference simulation. Fluxes through the reactions are in gigagrams Cl per year (Gg Cl yr−1). Heterogeneous reactions are represented in green. The main loss pathways for atomic chlorine are in orange. The numbers in the boxes represent the mass balances of the species families in Gg Cl.
In this section, the evolution of the model-calculated ozone sensitivity from the pre-industrial period to the present day is assessed, with and without halogen chemistry, and compared with results from other models. Then the impact of halogen chemistry on the sensitivity of ozone, OH radical and CH4 concentrations to the emissions of anthropogenic ozone precursors is discussed.
4.1 Pre-industrial to present-day changes
With and without halogen chemistry, the model simulates significantly lower ozone concentrations in the pre-industrial compared to present due to the decrease in NOx concentrations. Taking into account halogen chemistry reduces the average surface ozone concentrations for pre-industrial conditions by 5.0 ppbv (38 %), with the largest reductions over the oceans (5.5 ppbv or 42.4 %).
Globally, halogens reduce the tropospheric ozone burden by 67.1 Tg in the present day and 48.2 Tg in pre-industrial conditions (Table 7). Between PI and PD, ozone increases by 98.1 Tg (113 Tg in Sherwen et al., 2017) without halogen chemistry and by 79.2 Tg (90 Tg in Sherwen et al., 2017) with halogen chemistry. Both in PD and PI, halogens are responsible for about 23 %–28 % of the ozone destruction. Tropospheric ozone lifetimes drop from 21.1 to 17.9 d in the present with the inclusion of halogens and from 25.6 to 21.2 d in the pre-industrial period. The sensitivity of the burden of ground-level ozone to pre-industrial–present changes is ∼20 % lower when the chemistry of the halogens is considered. The changes observed in LMDZ-INCA and schematised in Fig. 7 are in agreement with the changes in Sherwen et al. (2017).
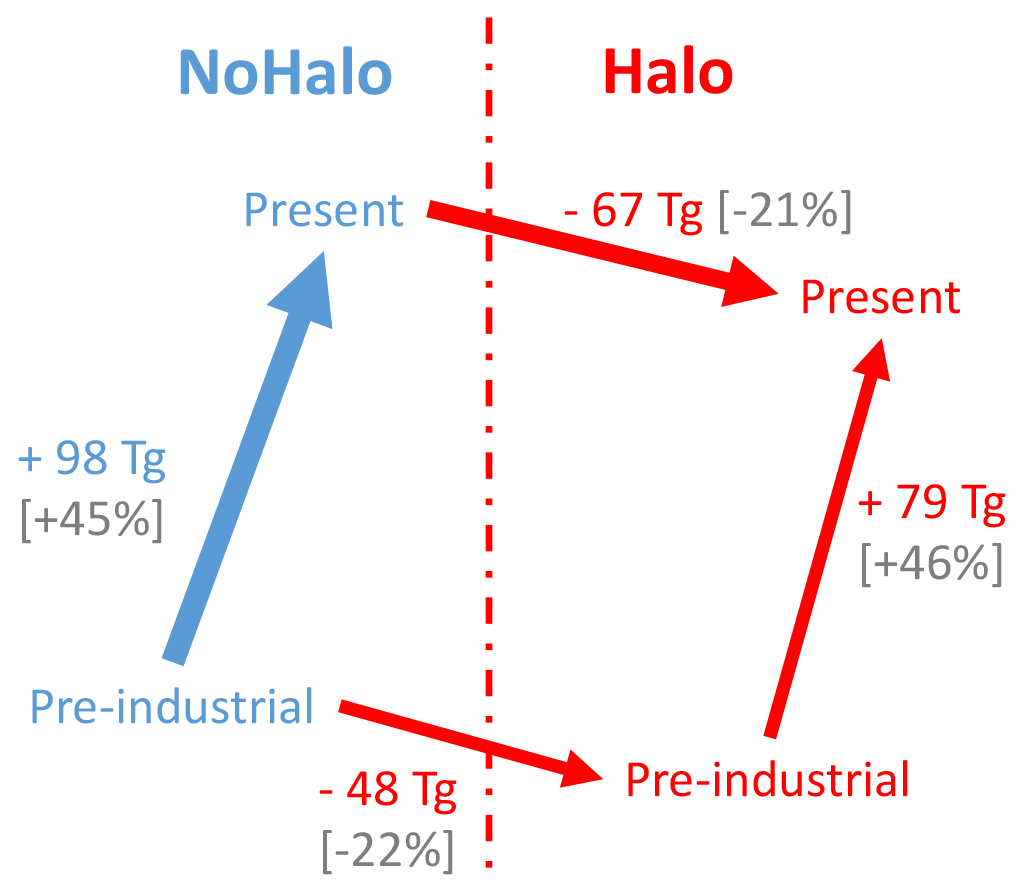
Figure 7Schematic representation of the evolution of ozone mass budget changes between PI and PD, following the inclusion of halogen chemistry in LMDZ-INCA.
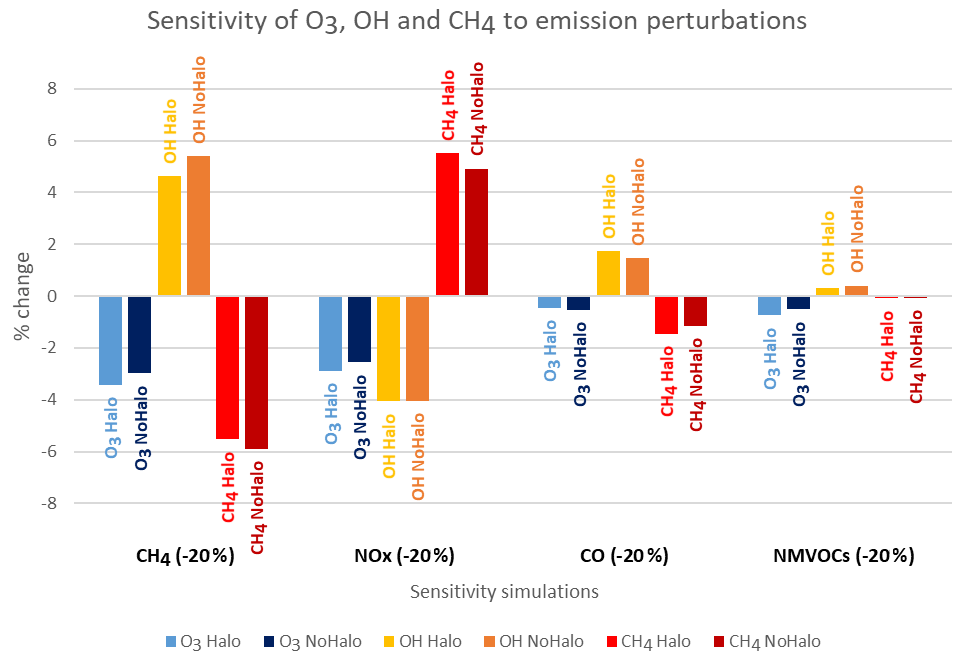
Figure 8Tropospheric relative changes (%) in ozone mass budget, OH concentration and methane lifetime for perturbation of NOx, CO and NMVOC emissions and CH4 concentrations with and without halogen chemistry.
4.2 Sensitivity runs
In order to study the effect of halogen chemistry on the sensitivity of the tropospheric oxidative system, we explore, in Fig. 8, the relative changes (%) in ozone and OH when present-day emissions of photo-oxidant precursors (NOx, CO and NMVOCs and of CH4 concentrations) are perturbed by a 20 % reduction, as done in the HTAP protocol. Our results show that the importance of sensitivities of ozone and OH to individual photo-oxidant precursor emissions increases as follows in terms of importance: NMVOCs, CO, NOx and CH4 (Fig. 8). It is also clear that the OH radical is more sensitive to perturbations in CH4, NOx and CO than ozone. Notably, when accounting for halogens, ozone becomes more sensitive to changes in CH4 and NOx and to a lesser extent to changes in NMVOCs. On the other hand, in the presence of halogen chemistry, the OH radical becomes more resilient in the presence of halogens to CH4, NOx and NMVOCs perturbations. Usually, solely the CH4 consumption by OH is considered when the chemical lifetime of CH4 in the troposphere is reported in the literature since it represents the main CH4 sink. It thus provides a useful proxy for global tropospheric oxidising capacity (Wild et al., 2020). The simulations examined here show that the CH4 lifetime with respect to OH remains a useful proxy for global tropospheric oxidising capacity for all perturbations. However, in the presence of halogen chemistry, when NOx emissions are reduced, atomic chlorine, a secondary oxidant of methane, becomes more efficient and increases the sensitivity of CH4 to NOx emission change. This exercise shows for the first time the influence of halogens on the sensitivity of the tropospheric oxidative system to changes in the emissions of photo-oxidant precursor (NOx, CO and NMVOCs and CH4).
4.3 Impact on methane lifetime
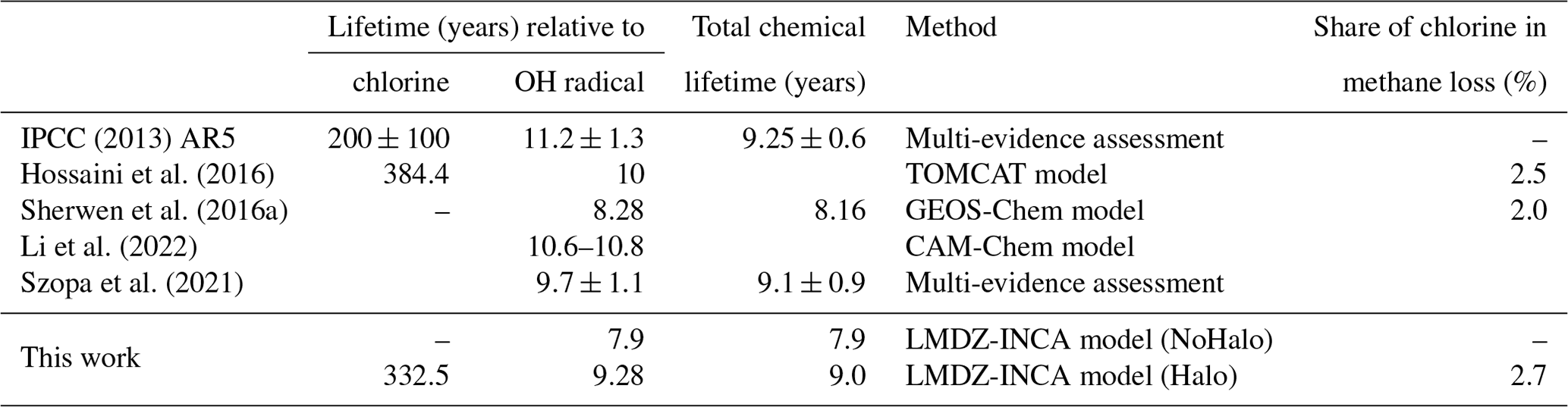
The inclusion of tropospheric halogens affects the atmospheric concentrations of a large number of compounds in the model. A marked decrease in oxidants (O3, OH, HO2, H2O2) is then computed. This leads to an increase in the concentrations of CO (+8.5 %) and some NMVOCs, including isoprene (+7.1 %). The lifetime of methane with respect to chlorine and the OH radical, as well as the share of chlorine in the losses of methane in the literature, is shown in Table 8. In the reference simulation, the tropospheric chemical lifetime of methane due to its consumption by OH is 7.9 years. With the inclusion of halogen chemistry, the OH concentration decreases, increasing this chemical lifetime of methane to 9.3 years. The chemical lifetime of methane linked to oxidation by Cl is 333 years, less than the 384 years suggested by Hossaini et al. (2016) but closer to IPCC Fifth Assessment Report (AR5) estimates. This oxidation by Cl represents 2.7 % of the total loss (close to the values reported in the literature and in Table 8). Thus, in LMDZ-INCA, accounting for both OH and the chlorinated radical consumption, methane oxidation leads to a total chemical lifetime of CH4 of 9.0 years.
Table 8Methane lifetime with respect to chlorine and OH radicals as well as the share of chlorine in methane losses in the literature.
Global chemistry climate models allow us to simulate the evolution of the atmospheric composition and are used for future projections in the framework of international model intercomparisons such as AerChemMIP. Most of these models do not account for halogen chemistry. We have implemented a simplified representation of the halogenated chemistry in the three-dimensional climate–chemistry model LMDZ-INCA to account for its effects on the tropospheric chemistry, notably ozone.
First, the halogenated species, their sources and sinks, their reactions in the gas and heterogeneous phases, and their physicochemical characteristics have been integrated into INCA. In order to simplify the integration of this chemistry, a chemical scheme already tested and evaluated in the CAM-Chem model has been used as a basis for our developments.
The representation of this chemistry has been shown to be sufficient in a first approach to simulate properly the impact of halogens on the photo-oxidising system in the troposphere and particularly in the atmospheric boundary layer. The response of the tropospheric chemical system Ox, HOx, NOx, CH4 and NMVOCs to the incorporation of halogen chemistry has been quantified. On a global tropospheric scale, this chemistry induces decreases in the ozone burden by 22 %, in OH by 8 % and in NOx by 33 %. An increase in CO and NMVOC concentrations is simulated, and the lifetime of methane increases by 1 year. These changes are consistent with those reported in the literature. Comparisons between simulations considering pre-industrial and present-day emissions show that the sensitivity of the ozone burden to changes in emissions over the pre-industrial era is ∼20 % lower in the presence of halogen chemistry (mainly because of the chemistry of iodine).
Sensitivity tests, consisting of reducing in turn the emissions of ozone precursors and methane concentrations, show for the first time that ozone becomes more sensitive to NOx, CO and NMVOC perturbations in the presence of halogen chemistry.
Although still rarely represented in chemistry climate models, our model results confirm that the chemistry of tropospheric halogens (Cl, Br, I) plays a significant role in the global loss of tropospheric ozone. Since halogen chemistry affects substantially the ozone burden both in pre-industrial and present-day chemical conditions and the ozone sensitivity to changes in anthropogenic emissions, international exercises examining the evolution of ozone between pre-industrial, present and future periods should consider taking into account halogen chemistry.
The main model codes and the chemical scheme with halogenated compounds as well as the data outputs are publicly accessible through https://doi.org/10.14768/56b1b070-deae-4357-a2dd-d75d27659f63 (Szopa et al., 2023).
The supplement related to this article is available online at: https://doi.org/10.5194/gmd-16-4041-2023-supplement.
The implementation of the halogens chemical scheme has been done by CC, SS and AC. The design of the numerical simulations was conducted by CC, SS, ASL and CAC. CC and SS prepared the figures. CC, SS, SB, ASL and CAC analysed the results and drafted the paper.
At least one of the (co-)authors is a member of the editorial board of Geoscientific Model Development. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The authors are thankful to the NCAR Atmospheric Chemistry Division (ACD) for the distribution of the NCAR/ACD TUV: Tropospheric Ultraviolet and Visible Radiation Model (https://www.acom.ucar.edu/Models/TUV/Interactive_TUV/, last access: 5 July 2023) and the availability of their quick tool.
This research has been supported by the Agence Nationale de la Recherche (grant no. ANR-16-CE31-0010) and the HPC resources of TGCC under the allocation A0090102212 made by GENCI (Grand Equipement National de Calcul Intensif).
This paper was edited by Samuel Remy and reviewed by six anonymous referees.
Abbatt, J. P. D., Thomas, J. L., Abrahamsson, K., Boxe, C., Granfors, A., Jones, A. E., King, M. D., Saiz-Lopez, A., Shepson, P. B., Sodeau, J., Toohey, D. W., Toubin, C., von Glasow, R., Wren, S. N., and Yang, X.: Halogen activation via interactions with environmental ice and snow in the polar lower troposphere and other regions, Atmos. Chem. Phys., 12, 6237–6271, https://doi.org/10.5194/acp-12-6237-2012, 2012.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I – gas phase reactions of Ox, HOx, NOx and SOx species, Atmos. Chem. Phys., 4, 1461–1738, https://doi.org/10.5194/acp-4-1461-2004, 2004.
Atkinson, R., Baulch, D. L., Cox. R. A., Crowley, J. N., Hampson Jr., R. F., Hynes, R. G., Jenkin, M. E., Kerr, J. A., Rossi, M. J., and Troe, J.: Summary of evaluated kinetic and photochemical data for atmospheric chemistry: web version February 2006, http://www.iupac-kinetic.ch.cam.ac.uk (last access: 13 July 2023), 2006.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume III – gas phase reactions of inorganic halogens, Atmos. Chem. Phys., 7, 981–1191, https://doi.org/10.5194/acp-7-981-2007, 2007.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume IV – gas phase reactions of organic halogen species, Atmos. Chem. Phys., 8, 4141–4496, https://doi.org/10.5194/acp-8-4141-2008, 2008.
Badia, A., Reeves, C. E., Baker, A. R., Saiz-Lopez, A., Volkamer, R., Koenig, T. K., Apel, E. C., Hornbrook, R. S., Carpenter, L. J., Andrews, S. J., Sherwen, T., and von Glasow, R.: Importance of reactive halogens in the tropical marine atmosphere: a regional modelling study using WRF-Chem, Atmos. Chem. Phys., 19, 3161–3189, https://doi.org/10.5194/acp-19-3161-2019, 2019.
Badia, A., Iglesias-Suarez, F., Fernandez, R. P., Cuevas, C. A., Kinnison, D. E., Lamarque, J.-F., Griffiths, P. T., Tarasick, D. W., Liu, J., and Saiz-Lopez, A.: The role of natural halogens inglobal tropospheric ozone chemistryand budget under different 21stcentury climate scenarios, J. Geophys. Res.-Atmos., 126, e2021JD034859, https://doi.org/10.1029/2021JD034859, 2021.
Barker, J. R., Steiner, A. L., and Wallington, T. J.: Advances In Atmospheric Chemistry, Volume 1, World Scientific Publishing Company, Adv. Atmos. Chem., World Scientific, ISBN 9789813271821, 2016.
Bedjanian, Y., Le Bras, G., and Poulet, G.: Kinetic study of the Br + IO, I + BrO and Br + I2 reactions. Heat of forma- tion of the BrO radical, Chem. Phys. Lett., 266, 233–238, https://doi.org/10.1016/S0009-2614(97)01530-3, 1997.
Bloss, W. J., Camredon, M., Lee, J. D., Heard, D. E., Plane, J. M. C., Saiz-Lopez, A., Bauguitte, S. J.-B., Salmon, R. A., and Jones, A. E.: Coupling of HOx, NOx and halogen chemistry in the antarctic boundary layer, Atmos. Chem. Phys., 10, 10187–10209, https://doi.org/10.5194/acp-10-10187-2010, 2010.
Bottenheim, J. W., Barrie, L. A., Atlas, E., Heidt, L. E., Niki, H., Rasmussen, R. A., and Shepson, P. B.: Depletion of lower tropospheric ozone during Arctic spring: The Polar Sunrise Experiment 1988, J. Geophys. Res.-Atmos., 95, 18555–18568, https://doi.org/10.1029/JD095iD11p18555, 1990.
Brasseur, G. P. and Solomon, S.: Aeronomy of the middle atmosphere: Chemistry and physics of the stratosphere and mesosphere (Vol. 32), Springer Science & Business Media, 2005.
Carpenter, L. J., Reimann, S., Burkholder, J. B., Clerbaux, C., Hall, B. D., Hossaini, R., Laube, J. C., and Yvon-Lewis, S. A.: Ozone-Depleting Substances (ODSs) and Other Gases of Interest to the Montreal Protocol, Chapter 1 in Scientific Assessment of Ozone Depletion: 2014, Global Ozone Research and Monitoring Project – Report No. 55, World Meteorological Organization, Geneva, Switzerland, 2014.
Carpenter, L. J.: Iodine in the Marine Boundary Layer, Chem. Rev., 103, 4953–4962, https://doi.org/10.1021/cr0206465, 2003.
Carpenter, L. J., MacDonald, S. M., Shaw, M. D., Kumar, R., Saunders, R. W., Parthipan, R., Wilson, J., and Plane, J. M. C.: Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine, Nat. Geosci., 6, 108–111, https://doi.org/10.1038/ngeo1687, 2013.
Chameides, W. L. and Davis, D. D.: Iodine: Its possible role in tropospheric photochemistry, J. Geophys. Res.-Ocean., 85, 7383–7398, https://doi.org/10.1029/JC085iC12p07383, 1980.
Cicerone, R. J.: Halogens in the atmosphere, Rev. Geophys., 19, 123–139, https://doi.org/10.1029/RG019i001p00123, 1981.
Dorf, M., Butz, A., Camy-Peyret, C., Chipperfield, M. P., Kritten, L., and Pfeilsticker, K.: Bromine in the tropical troposphere and stratosphere as derived from balloon-borne BrO observations, Atmos. Chem. Phys., 8, 7265–7271, https://doi.org/10.5194/acp-8-7265-2008, 2008.
Eastham, S. D., Weisenstein, D. K., and Barrett, S. R. H.: Development and evaluation of the unified tropospheric–stratospheric chemistry extension (UCX) for the global chemistry-transport model GEOS-Chem, Atmos. Environ., 89, 52–63, https://doi.org/10.1016/j.atmosenv.2014.02.001, 2014.
Fernandez, R. P., Barrera, J. A., López-Noreña, A. I., Kinnison, D. E., Nicely, J., Salawitch, R. J., Wales, P. A., Toselli, B. M., Tilmes, S., Lamarque, J.-F., Cuevas, C. A., and Saiz-Lopez, A.: Intercomparison between surrogate, explicit and full treatments of VSL bromine chemistry within the CAM-Chem chemistry-climate model, Geophys. Res. Lett., 48, e2020GL091125, https://doi.org/10.1029/2020GL091125, 2021.
Fernandez, R. P., Salawitch, R. J., Kinnison, D. E., Lamarque, J.-F., and Saiz-Lopez, A.: Bromine partitioning in the tropical tropopause layer: implications for stratospheric injection, Atmos. Chem. Phys., 14, 13391–13410, https://doi.org/10.5194/acp-14-13391-2014, 2014.
Fiore, A. M., Dentener, F. J., Wild, O., Cuvelier, C., Schultz, M. G., Hess, P., Textor, C., Schulz, M., Doherty, R. M., Horowitz, L. W., MacKenzie, I. A., Sanderson, M. G., Shindell, D. T., Stevenson, D. S., Szopa, S., Van Dingenen, R., Zeng, G., Atherton, C., Bergmann, D., Bey, I., Carmichael, G., Collins, W. J., Duncan, B. N., Faluvegi, G., Folberth, G., Gauss, M., Gong, S., Hauglustaine, D., Holloway, T., Isaksen, I. S. A., Jacob, D. J., Jonson, J. E., Kaminski, J. W., Keating, T. J., Lupu, A., Marmer, E., Montanaro, V., Park, R. J., Pitari, G., Pringle, K. J., Pyle, J. A., Schroeder, S., Vivanco, M. G., Wind, P., Wojcik, G., Wu, S., and Zuber, A.: Multimodel estimates of intercontinental source-receptor relationships for ozone pollution, J. Geophys. Res., 114, D04301, https://doi.org/10.1029/2008JD010816, 2009.
Folberth, G. A., Hauglustaine, D. A., Lathière, J., and Brocheton, F.: Interactive chemistry in the Laboratoire de Météorologie Dynamique general circulation model: model description and impact analysis of biogenic hydrocarbons on tropospheric chemistry, Atmos. Chem. Phys., 6, 2273–2319, https://doi.org/10.5194/acp-6-2273-2006, 2006.
Giorgi, F. and Chameides, W. L.: The rainout parameterization in a photochemical model, J. Geophys. Res.-Atmos., 90, 7872–7880, https://doi.org/10.1029/JD090iD05p07872, 1985.
Gomez Martin, J. C., Spietz, P., and Burrows, J.: Spectroscopic studies of the I2/O3 photochemistry: Part 1: Determination of the absolute absorption cross sections of iodine oxides of atmospheric relevance, J. Photochem. Photobiol. A, 176, 15–38, https://doi.org/10.1016/j.jphotochem.2005.09.024, 2005.
Gómez Martín, J. C., Spietz, P., and Burrows, J. P.: Kinetic and Mechanistic Studies of the I2/O3 Photochemistry, J. Phys. Chem. A, 111, 306–320, https://doi.org/10.1021/jp061186c, 2007.
Gómez Martín, J. C., Lewis, T. R., Blitz, M. A., Plane, J. M. C., Kumar, M., Francisco, J. S., and Saiz-Lopez, A.: A gas-to-particle conversion mechanism helps to explain atmospheric particle formation through clustering of iodine oxides, Nat. Commun., 11, 4521, https://doi.org/10.1038/s41467-020-18252-8, 2020.
Graedel, T. E. and Keene, W. C.: Tropospheric budget of reactive chlorine, Global Biogeochem. Cycles, 9, 47–77, https://doi.org/10.1029/94GB03103, 1995.
Hauglustaine, D. A., Hourdin, F., Jourdain, L., Filiberti, M., Walters, S., Lamarque, J., and Holland, E. A.: Interactive chemistry in the Laboratoire de Météorologie Dynamique general circulation model: Description and background tropospheric chemistry evaluation, J. Geophys. Res., 109, D04314, https://doi.org/10.1029/2003JD003957, 2004.
Hoesly, R. M., Smith, S. J., Feng, L., Klimont, Z., Janssens-Maenhout, G., Pitkanen, T., Seibert, J. J., Vu, L., Andres, R. J., Bolt, R. M., Bond, T. C., Dawidowski, L., Kholod, N., Kurokawa, J.-I., Li, M., Liu, L., Lu, Z., Moura, M. C. P., O'Rourke, P. R., and Zhang, Q.: Historical (1750–2014) anthropogenic emissions of reactive gases and aerosols from the Community Emissions Data System (CEDS), Geosci. Model Dev., 11, 369–408, https://doi.org/10.5194/gmd-11-369-2018, 2018.
Holmes, C. D., Prather, M. J., Søvde, O. A., and Myhre, G.: Future methane, hydroxyl, and their uncertainties: key climate and emission parameters for future predictions, Atmos. Chem. Phys., 13, 285–302, https://doi.org/10.5194/acp-13-285-2013, 2013.
Hossaini, R., Chipperfield, M. P., Montzka, S. A., Rap, A., Dhomse, S., and Feng, W.: Efficiency of short-lived halogens at influencing climate through depletion of stratospheric ozone, Nat. Geosci., 8, 186–190, https://doi.org/10.1038/ngeo2363, 2015.
Hossaini, R., Chipperfield, M. P., Saiz-Lopez, A., Fernandez, R., Monks, S., Feng, W., Brauer, P., and von Glasow, R.: A global model of tropospheric chlorine chemistry: Organic versus inorganic sources and impact on methane oxidation, J. Geophys. Res.-Atmos., 121, 14271–14297, https://doi.org/10.1002/2016JD025756, 2016.
Hourdin, F., Foujols, M.-A., Codron, F., Guemas, V., Dufresne, J.-L., Bony, S., Denvil, S., Guez, L., Lott, F., Ghattas, J., Braconnot, P., Marti, O., Meurdesoif, Y., and Bopp, L.: Impact of the LMDZ atmospheric grid configuration on the climate and sensitivity of the IPSL-CM5A coupled model, Clim. Dynam., 40, 2167–2192, https://doi.org/10.1007/s00382-012-1411-3, 2013.
Hu, L.: The Role of the Ocean in the Atmospheric Budgets of Methyl Bromide, Methyl Chloride and Methane. Doctoral dissertation, Texas A&M University, https://hdl.handle.net/1969.1/ETD-TAMU-2012-08-11592 (last access: 5 July 2023), 2012.
Iglesias-Suarez, F., Badia, A., Fernandez, R. P., Cuevas, C. A., Kinnison, D. E., Tilmes, S., Lamarque, J.-F., Long, M. C., Hossaini, R., and Saiz-Lopez, A.: Natural halogens buffer tropospheric ozone in a changing climate, Nat. Clim. Chang., 10, 147–154, https://doi.org/10.1038/s41558-019-0675-6, 2020.
IPCC (Intergovernmental Panel on Climate Change) (Ed.): Anthropogenic and natural radiative forcing, in Climate Change 2013 the Physical Science Basis: Working Group I Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, 9781107057, 659–740, 2013.
Kaltsoyannis, N. and Plane, J. M. C.: Quantum chemical calcula- tions on a selection of iodine-containing species (IO, OIO, INO3, (IO)2, I2O3, I2O4 and I2O5) of importance in the atmosphere, Phys. Chem. Chem. Phys., 10, 1723–1733, 2008.
King, K. D., Golden, D. M., and Benson, S. W.: Kinetics of the gas-phase thermal bromination of acetone. Heat of formation and stabilization energy of the acetonyl radical, J. Am. Chem. Soc., 92, 5541–5546, https://doi.org/10.1021/ja00722a001, 1970.
Lewis, T. R., Gómez Martín, J. C., Blitz, M. A., Cuevas, C. A., Plane, J. M. C., and Saiz-Lopez, A.: Determination of the absorption cross sections of higher-order iodine oxides at 355 and 532 nm, Atmos. Chem. Phys., 20, 10865–10887, https://doi.org/10.5194/acp-20-10865-2020, 2020.
Li, Q., Fernandez, R. P., Hossaini, R., Iglesias-Suarez, F., Cuevas, C. A., Apel, E. C., Kinnison, D. E., Lamarque, J. F., and Saiz-Lopez, A.: Reactive halogens increase the global methane lifetime and radiative forcing in the 21st century, Nat Commun., 13, 2768, https://doi.org/10.1038/s41467-022-30456-8, 2022.
Lobert, J. M., Keene, W. C., Logan, J. A., and Yevich, R.: Global chlorine emissions from biomass burning: Reactive Chlorine Emissions Inventory, J. Geophys. Res.-Atmos., 104, 8373–8389, https://doi.org/10.1029/1998JD100077, 1999.
Long, M. S., Keene, W. C., Easter, R. C., Sander, R., Liu, X., Kerkweg, A., and Erickson, D.: Sensitivity of tropospheric chemical composition to halogen-radical chemistry using a fully coupled size-resolved multiphase chemistry–global climate system: halogen distributions, aerosol composition, and sensitivity of climate-relevant gases, Atmos. Chem. Phys., 14, 3397–3425, https://doi.org/10.5194/acp-14-3397-2014, 2014.
Lurmann, F. W., Lloyd, A. C., and Atkinson, R.: A chemical mechanism for use in long-range transport/acid deposition computer modeling, J. Geophys. Res., 91, 10905–10936, https://doi.org/10.1029/JD091iD10p10905, 1986.
MacDonald, S. M., Gómez Martín, J. C., Chance, R., Warriner, S., Saiz-Lopez, A., Carpenter, L. J., and Plane, J. M. C.: A laboratory characterisation of inorganic iodine emissions from the sea surface: dependence on oceanic variables and parameterisation for global modelling, Atmos. Chem. Phys., 14, 5841–5852, https://doi.org/10.5194/acp-14-5841-2014, 2014.
McCulloch, A., Aucott, M. L., Benkovitz, C. M., Graedel, T. E., Kleiman, G., Midgley, P. M., and Li, Y.-F.: Global emissions of hydrogen chloride and chloromethane from coal combustion, incineration and industrial activities: Reactive Chlorine Emissions Inventory, J. Geophys. Res.-Atmos., 104, 8391–8403, https://doi.org/10.1029/1999JD900025, 1999.
McFiggans, G., Plane, J. M. C., Allan, B. J., Carpenter, L. J., Coe, H., and O'Dowd, C.: A modeling study of iodine chemistry in the marine boundary layer, J. Geophys. Res.-Atmos., 105, 14371–14385, https://doi.org/10.1029/1999JD901187, 2000.
Molina, M. J. and Rowland, F. S.: Stratospheric sink for chlorofluoromethanes: chlorine atom-catalysed destruction of ozone, Nature, 249, 810–812, https://doi.org/10.1038/249810a0, 1974.
Montzka, S. A., Reimannander, S., Engel, A., Kruger, K., O Doherty, S., Sturges, W. T., Blake, D. R., Dorf, M., Fraser, P. J., Froidevaux, L., Jucks, K., Kreher, K., Kurylo III, M. J., Mellouki, A., and Jo, D. P. V.: Ozone-Depleting Substances (ODSs) and Related Chemicals, Chapter 1 in Scientific Assessment of Ozone Depletion: 2010, Global Ozone Research and Monitoring Project – Report No. 52, World Meteorological Organization, Geneva, Switzerland, 2010.
Naik, V., Voulgarakis, A., Fiore, A. M., Horowitz, L. W., Lamarque, J.-F., Lin, M., Prather, M. J., Young, P. J., Bergmann, D., Cameron-Smith, P. J., Cionni, I., Collins, W. J., Dalsøren, S. B., Doherty, R., Eyring, V., Faluvegi, G., Folberth, G. A., Josse, B., Lee, Y. H., MacKenzie, I. A., Nagashima, T., van Noije, T. P. C., Plummer, D. A., Righi, M., Rumbold, S. T., Skeie, R., Shindell, D. T., Stevenson, D. S., Strode, S., Sudo, K., Szopa, S., and Zeng, G.: Preindustrial to present-day changes in tropospheric hydroxyl radical and methane lifetime from the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP), Atmos. Chem. Phys., 13, 5277–5298, https://doi.org/10.5194/acp-13-5277-2013, 2013.
O'Dowd, C. D., Jimenez, J. L., Bahreini, R., Flagan, R. C., Seinfeld, J. H., Hämeri, K., Pirjola, L., Kulmala, M., Jennings, S. G., and Hoffmann, T.: Marine aerosol formation from biogenic iodine emissions., Nature, 417, 632–636, https://doi.org/10.1038/nature00775, 2002.
Oltmans, S. J. and Komhyr, W. D.: Surface ozone distributions and variations from 1973–1984: Measurements at the NOAA Geophysical Monitoring for Climatic Change Baseline Observatories, J. Geophys. Res.-Atmos., 91, 5229–5236, https://doi.org/10.1029/JD091iD04p05229, 1986.
Ordóñez, C., Lamarque, J.-F., Tilmes, S., Kinnison, D. E., Atlas, E. L., Blake, D. R., Sousa Santos, G., Brasseur, G., and Saiz-Lopez, A.: Bromine and iodine chemistry in a global chemistry-climate model: description and evaluation of very short-lived oceanic sources, Atmos. Chem. Phys., 12, 1423–1447, https://doi.org/10.5194/acp-12-1423-2012, 2012.
Orlando, J. J. and Tyndall, G. S.: Rate Coefficients for the Thermal Decomposition of BrONO2 and the Heat of For- mation of BrONO2, J. Phys. Chem.-US, 100, 19398–19405, https://doi.org/10.1021/jp9620274, 1996.
Parrella, J. P., Jacob, D. J., Liang, Q., Zhang, Y., Mickley, L. J., Miller, B., Evans, M. J., Yang, X., Pyle, J. A., Theys, N., and Van Roozendael, M.: Tropospheric bromine chemistry: implications for present and pre-industrial ozone and mercury, Atmos. Chem. Phys., 12, 6723–6740, https://doi.org/10.5194/acp-12-6723-2012, 2012.
Platt, U. and Hönninger, G.: The role of halogen species in the troposphere., Chemosphere, 52, 325–338, https://doi.org/10.1016/S0045-6535(03)00216-9, 2003.
Prados-Roman, C., Cuevas, C. A., Fernandez, R. P., Kinnison, D. E., Lamarque, J.-F., and Saiz-Lopez, A.: A negative feedback between anthropogenic ozone pollution and enhanced ocean emissions of iodine, Atmos. Chem. Phys., 15, 2215–2224, https://doi.org/10.5194/acp-15-2215-2015, 2015a.
Prados-Roman, C., Cuevas, C. A., Hay, T., Fernandez, R. P., Mahajan, A. S., Royer, S.-J., Galí, M., Simó, R., Dachs, J., Großmann, K., Kinnison, D. E., Lamarque, J.-F., and Saiz-Lopez, A.: Iodine oxide in the global marine boundary layer, Atmos. Chem. Phys., 15, 583–593, https://doi.org/10.5194/acp-15-583-2015, 2015b.
Pszenny, A. A. P., Keene, W. C., Jacob, D. J., Fan, S., Maben, J. R., Zetwo, M. P., Springer-Young, M., Galloway, J. N.: Evidence of inorganic chlorine gases other than hydrogen chloride in marine surface air, Geophys. Res. Lett., 20, 699–702, https://doi.org/10.1029/93GL00047, 1993.
Riffault, V., Bedjanian, Y., and Poulet, G.: Kinetic and mechanistic study of the reactions of OH with IBr and HOI, J. Photoch. Photobiol. A, 176, 155–161, https://doi.org/10.1016/j.jphotochem.2005.09.002, 2005.
Sadourny, R. and Laval, K.: New Perspectives in Climate Modelling, edited by: Berger, A., 173–198, Elsevier, Amsterdam, 1984.
Saiz-Lopez, A. and Plane, J. M. C.: Novel iodine chemistry in the marine boundary layer, Geophys. Res. Lett., 31, L04112, https://doi.org/10.1029/2003GL019215, 2004.
Saiz-lopez, A. and Von Glasow, R.: Reactive halogen chemistry in the troposphere, Chem. Soc. Rev., 41, 6448–6472, https://doi.org/10.1039/c2cs35208g, 2012.
Saiz-Lopez, A., Lamarque, J.-F., Kinnison, D. E., Tilmes, S., Ordóñez, C., Orlando, J. J., Conley, A. J., Plane, J. M. C., Mahajan, A. S., Sousa Santos, G., Atlas, E. L., Blake, D. R., Sander, S. P., Schauffler, S., Thompson, A. M., and Brasseur, G.: Estimating the climate significance of halogen-driven ozone loss in the tropical marine troposphere, Atmos. Chem. Phys., 12, 3939–3949, https://doi.org/10.5194/acp-12-3939-2012, 2012a.
Saiz-Lopez, A., Plane, J. M. C., Baker, A. R., Carpenter, L. J., von Glasow, R., Gómez Martín, J. C., McFiggans, G., and Saunders, R. W.: Atmospheric Chemistry of Iodine, Chem. Rev., 112, 1773–1804, https://doi.org/10.1021/cr200029u, 2012b.
Saiz-Lopez, A., Fernandez, R. P., Ordóñez, C., Kinnison, D. E., Gómez Martín, J. C., Lamarque, J.-F., and Tilmes, S.: Iodine chemistry in the troposphere and its effect on ozone, Atmos. Chem. Phys., 14, 13119–13143, https://doi.org/10.5194/acp-14-13119-2014, 2014.
Saiz-Lopez, A., Baidar, S., Cuevas, C. A., Koenig, T. K., Fernandez, R. P., Dix, B., Kinnison, D. E., Lamarque, J.-F., Rodriguez-Lloveras, X., Campos, T. L., and Volkamer, R.: Injection of iodine to the stratosphere, Geophys. Res. Lett., 42, 6852–6859, https://doi.org/10.1002/2015GL064796, 2015.
Saltzman, E. S., Aydin, M., De Bruyn, W. J., King, D. B., and Yvon-Lewis, S. A.: Methyl bromide in preindustrial air: Measurements from an Antarctic ice core, J. Geophys. Res.-Atmos., 109, D05301, https://doi.org/10.1029/2003JD004157, 2004.
Sander, S., Friedl, R., Golden, D., Kurylo, M., Huie, R., Orkin, V., Moortgat, G., Ravishankara, A. R., Kolb, C., Molina, M., and Finlayson-Pitts, B.: Chemical Kinetics and Photochemical Data for Use in Atmosheric Studies; JPL Publication 02-25, 2003.
Sander, S. P., Abbatt, J. P. D., Barker, J. R., Burkholder, J. B., Friedl, R. R., Golden, D. M., Huie, R. E., Kolb, C. E., Kurylo, M. J., Moortgat, G. K., Orkin, V. L., and Wine, P. H.: Chemical kinetics and photochemical data for use in atmospheric studies: Evaluation number 17, Technical Report 2011, JPL Publication 10-6 , http://jpldataeval.jpl.nasa.gov/pdf/JPL 10-6 Final 15June2011.pdf (last access: 5 July 2023), 2011.
Schmidt, J. A., Jacob, D. J., Horowitz, H. M., Hu, L., Sherwen, T., Evans, M. J., Liang, Q., Suleiman, R. M., Oram, D. E., Le Breton, M., Percival, C. J., Wang, S., Dix, B., and Volkamer, R.: Modeling the observed tropospheric BrO background: Importance of multiphase chemistry and implications for ozone, OH, and mercury, J. Geophys. Res.-Atmos., 819–835, https://doi.org/10.1002/2015JD024229, 2016.
Seakins, P. W., Pilling, M. J., Niiranen, J. T., Gutman, D., and Krasnoperov, L. N.: Kinetics and thermochemistry of R + HBr RH + Br reactions: determinations of the heat of formation of C2H5, i-C3H7, sec-C4H9, and t-C4H9, J. Phys. Chem., 96, 9847–9855, 1992.
Sherwen, T., Evans, M. J., Carpenter, L. J., Andrews, S. J., Lidster, R. T., Dix, B., Koenig, T. K., Sinreich, R., Ortega, I., Volkamer, R., Saiz-Lopez, A., Prados-Roman, C., Mahajan, A. S., and Ordóñez, C.: Iodine's impact on tropospheric oxidants: a global model study in GEOS-Chem, Atmos. Chem. Phys., 16, 1161–1186, https://doi.org/10.5194/acp-16-1161-2016, 2016a.
Sherwen, T., Schmidt, J. A., Evans, M. J., Carpenter, L. J., Großmann, K., Eastham, S. D., Jacob, D. J., Dix, B., Koenig, T. K., Sinreich, R., Ortega, I., Volkamer, R., Saiz-Lopez, A., Prados-Roman, C., Mahajan, A. S., and Ordóñez, C.: Global impacts of tropospheric halogens (Cl, Br, I) on oxidants and composition in GEOS-Chem, Atmos. Chem. Phys., 16, 12239–12271, https://doi.org/10.5194/acp-16-12239-2016, 2016b.
Sherwen, T., Evans, M. J., Carpenter, L. J., Schmidt, J. A., and Mickley, L. J.: Halogen chemistry reduces tropospheric O3 radiative forcing, Atmos. Chem. Phys., 17, 1557–1569, https://doi.org/10.5194/acp-17-1557-2017, 2017.
Simpson, W. R., Brown, S. S., Glasow, R. Von, Saiz-lopez, A. and Thornton, J. A.: Tropospheric Halogen Chemistry: Sources, Cycling, and Impacts, Chem Rev., 115, 4035–4062, https://doi.org/10.1021/cr5006638, 2015.
Spietz, P., Gómez Martín, J. C., and Burrows, J. P.: Spectroscopic studies of the I2/O3 photochemistry: Part 2. Improved spectra of iodine oxides and analysis of the IO absorption spectrum, J. Photochem. Photobiol. A, 176, 50–67, https://doi.org/54510.1016/j.jphotochem.2005.08.023, 2005.
Stevenson, D. S., Zhao, A., Naik, V., O'Connor, F. M., Tilmes, S., Zeng, G., Murray, L. T., Collins, W. J., Griffiths, P. T., Shim, S., Horowitz, L. W., Sentman, L. T., and Emmons, L.: Trends in global tropospheric hydroxyl radical and methane lifetime since 1850 from AerChemMIP, Atmos. Chem. Phys., 20, 12905–12920, https://doi.org/10.5194/acp-20-12905-2020, 2020.
Stolarski, R. S. and Cicerone, R. J.: Stratospheric Chlorine: a Possible Sink for Ozone, Can. J. Chem., 52, 1610–1615, https://doi.org/10.1139/v74-233, 1974.
Stone, D., Whalley, L. K., and Heard, D. E.: Tropospheric OH and HO2 radicals: field measurements and model comparisons, Chem. Soc. Rev., 41, 6348–6404, https://doi.org/10.1039/C2CS35140D, 2012.
Szopa, S., Balkanski, Y., Schulz, M., Bekki, S., Cugnet, D., Fortems-Cheiney, A., Turquety, S., Cozic, A., Déandreis, C., Hauglustaine, D., Idelkadi, A., Lathière, J., Lefevre, F., Marchand, M., Vuolo, R., Yan, N., and Dufresne, J.-L.: Aerosol and ozone changes as forcing for climate evolution between 1850 and 2100, Clim. Dynam., 40, 2223–2250, https://doi.org/10.1007/s00382-012-1408-y, 2013.
Szopa, S., Naik, V., Adhikary, B., Artaxo, P., Berntsen, T., Collins, W. D., Fuzzi, S., Gallardo, L., Kiendler-Scharr, A., Klimont, Z., Liao, H., Unger, N., and Zanis, P.: Short-Lived Climate Forcers. In Climate Change 2021: The Physical Science Basis, Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, edited by: Masson-Delmotte, V., Zhai, P., Pirani, A., Connors, S. L., Péan, C., Berger, S., Caud, N., Chen, Y., Goldfarb, L., Gomis, M. I., Huang, M., Leitzell, K., Lonnoy, E., Matthews, J. B. R., Maycock, T. K., Waterfield, T., Yelekçi, O., Yu, R., and Zhou, B., Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 817–922, https://doi.org/10.1017/9781009157896.008, 2021.
Szopa, S., Cozic, A., and Caram, C.: INCA_HalogTropo_GMD_2023, IPSL [code and data], https://doi.org/10.14768/56b1b070-deae-4357-a2dd-d75d27659f63, 2023.
Thomas, J. L., Stutz, J., Lefer, B., Huey, L. G., Toyota, K., Dibb, J. E., and von Glasow, R.: Modeling chemistry in and above snow at Summit, Greenland – Part 1: Model description and results, Atmos. Chem. Phys., 11, 4899–4914, https://doi.org/10.5194/acp-11-4899-2011, 2011.
von Glasow, R., Sander, R., Bott, A., and Crutzen, P. J.: Modeling halogen chemistry in the marine boundary layer 1. Cloud-free MBL, J. Geophys. Res.-Atmos., 107, ACH 9-1–ACH 9-16, https://doi.org/10.1029/2001JD000942, 2002.
von Glasow, R., von Kuhlmann, R., Lawrence, M. G., Platt, U., and Crutzen, P. J.: Impact of reactive bromine chemistry in the troposphere, Atmos. Chem. Phys., 4, 2481–2497, https://doi.org/10.5194/acp-4-2481-2004, 2004.
Wang, X., Jacob, D. J., Eastham, S. D., Sulprizio, M. P., Zhu, L., Chen, Q., Alexander, B., Sherwen, T., Evans, M. J., Lee, B. H., Haskins, J. D., Lopez-Hilfiker, F. D., Thornton, J. A., Huey, G. L., and Liao, H.: The role of chlorine in global tropospheric chemistry, Atmos. Chem. Phys., 19, 3981–4003, https://doi.org/10.5194/acp-19-3981-2019, 2019.
Wang, X., Jacob, D. J., Downs, W., Zhai, S., Zhu, L., Shah, V., Holmes, C. D., Sherwen, T., Alexander, B., Evans, M. J., Eastham, S. D., Neuman, J. A., Veres, P. R., Koenig, T. K., Volkamer, R., Huey, L. G., Bannan, T. J., Percival, C. J., Lee, B. H., and Thornton, J. A.: Global tropospheric halogen (Cl, Br, I) chemistry and its impact on oxidants, Atmos. Chem. Phys., 21, 13973–13996, https://doi.org/10.5194/acp-21-13973-2021, 2021.
Wang, Y. and Jacob, D. J.: Anthropogenic forcing on tropospheric ozone and OH since preindustrial times, J. Geophys. Res.-Atmos., 103, 31123–31135, https://doi.org/10.1029/1998JD100004, 1998.
Wesely, M. L.: Parameterization of surface resistances to gaseous dry deposition in regional-scale numerical models, Atmos. Environ., 23, 1293–1304, https://doi.org/10.1016/0004-6981(89)90153-4, 1989.
Wild, O., Voulgarakis, A., O'Connor, F., Lamarque, J.-F., Ryan, E. M., and Lee, L.: Global sensitivity analysis of chemistry–climate model budgets of tropospheric ozone and OH: exploring model diversity, Atmos. Chem. Phys., 20, 4047–4058, https://doi.org/10.5194/acp-20-4047-2020, 2020.
Worton, D. R., Sturges, W. T., Schwander, J., Mulvaney, R., Barnola, J.-M., and Chappellaz, J.: 20th century trends and budget implications of chloroform and related tri-and dihalomethanes inferred from firn air, Atmos. Chem. Phys., 6, 2847–2863, https://doi.org/10.5194/acp-6-2847-2006, 2006.
Yang, X., Cox, R. A., Warwick, N. J., Pyle, J. A., Carver, G. D., O'Connor, F. M., and Savage, N. H.: Tropospheric bromine chemistry and its impacts on ozone: A model study, J. Geophys. Res.-Atmos., 110, https://doi.org/10.1029/2005JD006244, 2005.
Young, P. J., Archibald, A. T., Bowman, K. W., Lamarque, J.-F., Naik, V., Stevenson, D. S., Tilmes, S., Voulgarakis, A., Wild, O., Bergmann, D., Cameron-Smith, P., Cionni, I., Collins, W. J., Dalsøren, S. B., Doherty, R. M., Eyring, V., Faluvegi, G., Horowitz, L. W., Josse, B., Lee, Y. H., MacKenzie, I. A., Nagashima, T., Plummer, D. A., Righi, M., Rumbold, S. T., Skeie, R. B., Shindell, D. T., Strode, S. A., Sudo, K., Szopa, S., and Zeng, G.: Pre-industrial to end 21st century projections of tropospheric ozone from the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP), Atmos. Chem. Phys., 13, 2063–2090, https://doi.org/10.5194/acp-13-2063-2013, 2013.
Zhu, L., Jacob, D. J., Eastham, S. D., Sulprizio, M. P., Wang, X., Sherwen, T., Evans, M. J., Chen, Q., Alexander, B., Koenig, T. K., Volkamer, R., Huey, L. G., Le Breton, M., Bannan, T. J., and Percival, C. J.: Effect of sea salt aerosol on tropospheric bromine chemistry, Atmos. Chem. Phys., 19, 6497–6507, https://doi.org/10.5194/acp-19-6497-2019, 2019.
- Abstract
- Introduction
- Methodology
- Evaluation of the representation of the halogen-driven ozone sinks
- Perturbation of the ozone sensitivity to its precursors by halogen chemistry
- Conclusions
- Code and data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement
- Abstract
- Introduction
- Methodology
- Evaluation of the representation of the halogen-driven ozone sinks
- Perturbation of the ozone sensitivity to its precursors by halogen chemistry
- Conclusions
- Code and data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement