the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 24 Jun 2021
| 24 Jun 2021
Effects of heterogeneous reactions on tropospheric chemistry: a global simulation with the chemistry–climate model CHASER V4.0
Phuc T. M. Ha
Ryoki Matsuda
Yugo Kanaya
Fumikazu Taketani
Kengo Sudo
This study uses a chemistry–climate model CHASER (MIROC) to explore the roles of heterogeneous reactions (HRs) in global tropospheric chemistry. Three distinct HRs of N2O5, HO2, and RO2 are considered for surfaces of aerosols and cloud particles. The model simulation is verified with EANET and EMEP stationary observations; R/V Mirai ship-based data; ATom1 aircraft measurements; satellite observations by OMI, ISCCP, and CALIPSO-GOCCP; and reanalysis data JRA55. The heterogeneous chemistry facilitates improvement of model performance with respect to observations for NO2, OH, CO, and O3, especially in the lower troposphere. The calculated effects of heterogeneous reactions cause marked changes in global abundances of O3 (−2.96 %), NOx (−2.19 %), CO (+3.28 %), and global mean CH4 lifetime (+5.91 %). These global effects were contributed mostly by N2O5 uptake onto aerosols in the middle troposphere. At the surface, HO2 uptake gives the largest contributions, with a particularly significant effect in the North Pacific region (−24 % O3, +68 % NOx, +8 % CO, and −70 % OH), mainly attributable to its uptake onto clouds. The RO2 reaction has a small contribution, but its global mean negative effects on O3 and CO are not negligible. In general, the uptakes onto ice crystals and cloud droplets that occur mainly by HO2 and RO2 radicals cause smaller global effects than the aerosol-uptake effects by N2O5 radicals (+1.34 % CH4 lifetime, +1.71 % NOx, −0.56 % O3, +0.63 % CO abundances). Nonlinear responses of tropospheric O3, NOx, and OH to the N2O5 and HO2 uptakes are found in the same modeling framework of this study (R>0.93). Although all HRs showed negative tendencies for OH and O3 levels, the effects of HR(HO2) on the tropospheric abundance of O3 showed a small increment with an increasing loss rate. However, this positive tendency turns to reduction at higher rates (>5 times). Our results demonstrate that the HRs affect not only polluted areas but also remote areas such as the mid-latitude sea boundary layer and upper troposphere. Furthermore, HR(HO2) can bring challenges to pollution reduction efforts because it causes opposite effects between NOx (increase) and surface O3 (decrease).
- Article
(24396 KB) - Full-text XML
-
Supplement
(8008 KB) - BibTeX
- EndNote




Heterogeneous reactions (HRs) on the surfaces of atmospheric aerosols and cloud droplets are regarded as playing crucial roles in atmospheric chemistry. They affect ozone (O3) concentrations in various pathways via the cycle of odd hydrogen (HOx) and nitrogen oxides (NOx) (Jacob, 2000). Tropospheric ozone, an important greenhouse gas, causes damage to human health, crops, and ecosystem productivity (Monks et al., 2015). Although tropospheric O3 was recognized as a critical oxidant species, its global distribution has not been adequately captured to date because of the limited number of observations. Whereas many sites in the heavily polluted regions of eastern Asia show ozone increases since 2000 (Liu and Wang, 2020), many sites in other regions show decreases (Gaudel et al., 2018). Moreover, O3 responds to changes of multiple pollutants such as NOx and volatile organic compounds (VOCs) in different ways, which challenge the local pollutant control policy. For instance, since the Chinese government released the Air Pollution Prevention and Control Action Plan in 2010 (Zheng et al., 2018), the targets of SO2, NOx, and particulate matter (PM) decreased drastically, but urban ozone pollution has been worsening (Liu and Wang, 2020). Indeed, the O3 responses are controlled by several mechanisms, including heterogeneous effects of HO2 and N2O5 onto aerosols (Kanaya et al., 2009; Li et al., 2019; Liu and Wang, 2020; Taketani et al., 2012).
Stationary observations and laboratory experiments are important for enhancing the understanding of the tropospheric chemistry of O3 and other essential components (NOx, HOx). However, direct observation of vertical O3 distribution, including upper tropospheric O3, was not available before 1970. It has been deployed only at limited sites across the globe. Global atmospheric modeling is a useful method to reanalyze or forecast the past and future changes in O3 and their effects on human health and the climate. To serve this task, atmospheric models use both laboratory and observational data to help achieve accurate simulations of O3 and its precursors (HOx, NOx, hydrocarbons). To date, many modeling studies have suggested that heterogeneous chemistry be included in a standard model for tropospheric chemistry (Jacob, 2000; Macintyre and Evans, 2010, 2011; de Reus et al., 2005).
One fundamentally important HR in the troposphere is the uptake of N2O5 onto aqueous aerosols, known as a removal pathway for NOx at night (Platt et al., 1984). Actually, NOx plays crucially important roles in the troposphere because it controls the cycle of HOx and the production rate of tropospheric O3 (Logan et al., 1981; Riemer et al., 2003). The morning photochemistry can be affected by NO3 and N2O5, which are important nocturnal oxidants. Since the early 1980s, the role of urban NOx chemistry in Los Angeles pollution (National Research Council, 1991) has been acknowledged, but the proclamation of nighttime radicals remained sparse. It was only recognized in the past decade that N2O5 radical chemistry could have a much more perceptible effect stemming from reasons including a refined understanding of heterogeneous processes occurring at night (Brown and Stutz, 2012). The HR of N2O5 was revealed under different meteorological conditions in the US, Europe, and China (photosmog, high relative humidity (RH), or seasonal variation) for particles of various types: ice, aqueous aerosols with organic-coating, urban aerosols, dust, and soot (Apodaca et al., 2008; Lowe et al., 2015; Qu et al., 2019; Riemer et al., 2003, 2009; Wang et al., 2018, 2017; Xia et al., 2019). The uptake of N2O5 can markedly enhance nitrate concentration in nocturnal chemistry or PM2.5 explosive growth events in summer, decrease NOx, and either increase or decrease O3 concentrations in different NOx conditions (Dentener and Crutzen, 1993; Qu et al., 2019; Riemer et al., 2003; Wang et al., 2017). Even during daytime, N2O5 in the marine boundary layers can enhance the NOx to HNO3 conversion, and chemical destruction of O3 (Osthoff et al., 2006). A 10–20 ppbv reduction of O3 because of N2O5 uptake in the polluted regions of China has also been reported (Li et al., 2018). At mid- to high latitudes, N2O5 uptakes on sulfate aerosols could engender 80 % and 10 % NOx reduction, respectively, in winter and summer, leading to an approximate 10 % reduction of O3 in both seasons (Li et al., 2018).
Another vital process taking place on particles is the HRs of peroxy radicals (HO2 and RO2). Peroxy radicals are the primary chain carriers driving O3 production in the troposphere. Moreover, it can drive the hydrocarbon and NOx concentrations, which are important for nocturnal radical chemistry (Geyer et al., 2003; Richard, 2000; Salisbury et al., 2001). In the past, the HR(HO2) effects have been well considered in the laboratory (Macintyre and Evans, 2011) and field observations (Kanaya et al., 2001, 2002a, b, 2003, 2007; Taketani et al., 2012), but many technical problems (e.g., detecting HO2) have created difficulties that challenge its reported importance in the troposphere, as asserted from recent studies (Liao and Seinfeld, 2005; Martin et al., 2003; Tie et al., 2001). More recently, global modeling reports have described that the inclusion of HO2 uptake can affect atmospheric constituents strongly by the increment in tropospheric abundances for carbon monoxide (CO) and other trace gases because of reduced oxidation capacity (Lin et al., 2012; Macintyre and Evans, 2011). The HOx loss on aerosols can reduce O3 concentrations by up to 33 % in remote areas and up to 10 % in a smog episode (Saathoff et al., 2001; Taketani et al., 2012). The HOx loss on sea salt, sulfate, and organic carbon in various environments can decrease HO2 levels by 6 %–13 %, 10 %–40 %, and 40 %–70 %, respectively (Martin et al., 2003; Taketani et al., 2008, 2009; Tie et al., 2001). For RO2 with a typical representative of CH3CO.O2 (peroxyacetyl radical, PA), it plays a big role in the long-range transport of pollution (VOC, NOx) (Richard, 2000; Villalta et al., 1996). It can bring NOx from polluted domains as peroxyacyl nitrates (PAN) to remote regions in the ocean and higher altitudes (Qin et al., 2018; Richard, 2000). The concentrations of HO2 and RO2 at nighttime in the marine boundary layer were measured and confirmed (Geyer et al., 2003; Salisbury et al., 2001). Moreover, some evidence suggests uptake of HO2 and PA on clouds, aqueous aerosols, and other surfaces in high-humidity conditions, although the mechanism is uncertain (Geyer et al., 2003; Jacob, 2000; Kanaya et al., 2002b; Liao and Seinfeld, 2005; Lin et al., 2012; Richard, 2000; Salisbury et al., 2001). The predominance of peroxy uptake to clouds results from the ubiquitous existence and larger SAD maxima of cloud droplets in the atmosphere. Indeed, aqueous-phase chemistry might represent an important sink for O3 (Lelieveld and Crutzen, 1990). In addition, PA loss on aqueous particles can mediate the loss of PAN (CH3CO.O2NO) in fog (Villalta et al., 1996). Some modeling studies indicate that HOx loss (including HO2 loss) on aqueous aerosols reduces OH by 2 % , increases CO by 7 % and increases O3 by 0.5 % in the annual mean global burden (Huijnen et al., 2014). However, in a coastal environment in the Northern Hemisphere it increases OH by 15 % and reduces HO2 by 30 % (Sommariva et al., 2006; Thornton et al., 2008).
Although the contributions of each uptake category to tropospheric chemistry differ and must be considered both separately and as a whole, few studies have provided a global overview of heterogeneous chemistry the comprehensively examines the uptakes of N2O5, HO2, and RO2 on widely various particles. For instance, uptakes of both N2O5 and HO2 tend to reduce O3 in particular environments (Li et al., 2018; Saathoff et al., 2001; Taketani et al., 2012), but the HO2 loss on clouds can increase the tropospheric O3 burden (Huijnen et al., 2014). The latter trend is not widely suggested yet because the cloud chemistry is still neglected in many O3 models (Stadtler et al., 2018; Thornton et al., 2008). The predominant effects of HO2 uptake on aerosols compared to the effect by N2O5 were reported during the summer smog condition (Saathoff et al., 2001) but with lack of confirmation on a global scale. Moreover, the heterogeneous effects of RO2 have been investigated only insufficiently (Jacob, 2000). In this study, we examine these uncertainties using the global model CHASER to perceive the respective and total effects of the HRs of N2O5, HO2, and RO2 on the tropospheric chemistry. For the interface of HRs in the atmosphere, we tentatively consider surfaces of cloud particles and those of aerosols and discuss details of its effects in this study. In the following text, the research method, including model description and configuration, is described in Sect. 2. In Sect. 3.1, our model is verified with available observations including ground stations and ship, aircraft, and satellite measurements, particularly addressing the roles of the HRs. The global effects of N2O5, HO2, and RO2 uptake are discussed in Sect. 3.2 to elucidate cloud particles and aerosol effects. Section 3.3 will discuss sensitivities of tropospheric chemistry to the magnitudes of HRs. Section 4 presents a summary and concluding remarks.
2.1 Global chemistry model
The global chemistry model used for this study is CHASER (MIROC-ESM) (Sudo et al., 2002; Sudo and Akimoto, 2007; Watanabe et al., 2011), which considers detailed photochemistry in the troposphere and stratosphere. The chemistry component of the model, based on CHASER-V4.0, calculates the concentrations of 92 chemical species and 262 chemical reactions (58 photolytic, 183 kinetic, and 21 heterogeneous reactions including reactions on polar stratospheric clouds); more details on CHASER can be found in an earlier report of the literature (Morgenstern et al., 2017). Its tropospheric chemistry considers the fundamental chemical cycle of Ox–NOx–HOx–CH4–CO, along with oxidation of non-methane volatile organic compounds (NMVOCs). Its stratospheric chemistry simulates chlorine and bromine-containing compounds, CFCs, HFCs, carbonyl sulfide (OCS), NO2, and the formation of polar stratospheric clouds (PSCs) and heterogeneous reactions on PSC surfaces. In the framework of MIROC-Chem, CHASER is coupled with the MIROC-AGCM atmospheric general circulation model (version 4; Watanabe et al., 2011). The meteorological fields simulated by MIROC-AGCM were nudged toward the 6-hourly NCEP FNL data (https://rda.ucar.edu/datasets/ds083.2/, last access: 30 October 2018). For this study, the spatial resolution of the model was set as T42 (about 2.8∘ × 2.8∘ grid spacing) in horizontal and L36 (surface to approx. 50 km) in vertical. Anthropogenic emissions for O3 and aerosol precursors like NOx, CO, VOCs, and SO2 are specified using the HTAP-II inventory (Janssens-Maenhout et al., 2015), with biomass burning emissions derived from the MACC reanalysis system (Inness et al., 2013).
In the model, the aerosol concentrations for black carbon (BC) / organic carbon (OC), sea salt, and soil dust are handled by the SPRINTAR module, which is also based on the CCSR/NIES AGCM (Takemura et al., 2000). The bulk thermodynamics for aerosols are applied, including SO chemistry (SO2 oxidation with OH, O3/H2O2, which is cloud-pH dependent) SO–NO–NH and SO–dust interaction.
2.2 Heterogeneous reactions in the chemistry–climate model (CHASER)
The CHASER-V4 model considers HRs in both the troposphere and stratosphere. In this work, we particularly examine HRs in the troposphere. In the current version of CHASER, tropospheric HRs are considered for N2O5, HO2, and RO2, using uptake coefficients for the distinct surfaces of aerosols (sulfate, sea salt, dust, and organic carbons) and cloud particles (liquid/ice) as listed in Table 2. Although some other views incorporate the catalysis of transition metal ions (TMIs) Cu(I)/Cu(II) and Fe(II)/Fe(III) for the HO2 conversion on aqueous aerosols (Li et al., 2018; Mao et al., 2013; Taketani et al., 2012), this mechanism remains uncertain (Jacob, 2000). The TMI mechanism might lead to either H2O2 (Jacob, 2000) or H2O products (Mao et al., 2013). However, this may not cause any significant difference, since recycling HO2 from H2O2 is ineffective (Li et al., 2018). For this study, the uptake of HO2 is affirmed with H2O2 as the product (Loukhovitskaya et al., 2009; Taketani et al., 2009), as it is generally used in many atmospheric models such that this is not counted as a terminal sink for HO2 (Jacob, 2000; Lelieveld and Crutzen, 1990; Morita et al., 2004; Thornton et al., 2008). The RO2 uptakes are assumed with inert products, as suggested by Jacob (2000). The heterogeneous pseudo-first-order loss rate β for the species i is given using the theory of Schwartz (Dentener and Crutzen, 1993; Jacob, 2000; Schwartz, 1986), in which it is simply treated with the mass transfer limitations operating two conductances representing free molecular and continuum regimes for tropospheric clouds and aerosols, in addition to using reactive uptake coefficient (γ) instead of the mass accommodation coefficient as follows:
where νi stands for the mean molecular speed (cm s−1) of species i, Dij is the gaseous mass transfer (diffusion) coefficient (cm2 s−1) of species i for particle type j, and Aj expresses the surface area density (cm2 cm−3) for particle type j. In the model, the particle size and effective radius Rj for aerosols are calculated as a function of RH (Takemura et al., 2000). The aerosol concentrations are based on SPRINTAR for , sea salt, and dust (Takemura et al., 2000). The surface area density (SAD) for aerosols (Aj) is estimated using lognormal distributions of particle size (SFj) with mode radii variable with the RH (Sudo et al., 2002) as follows
where CN represents number density (cm−3) and Rj signifies the effective radii (cm) of particle type j. To calculate SAD for cloud particles, the liquid water content (LWC) and ice water content (IWC) in the AGCM are converted using the cloud droplet distribution of Battan and Reitan (1957) and the relation between IWC and the surface area density for ice clouds (Lawrence and Crutzen, 1998; McFarquhar and Heymsfield, 1996).
In these equations, Ac represents the cross-section area for ice crystals (cm2 cm−3). For liquid clouds, the following holds:
The uptake coefficient parameter (γ) is defined as the net probability that a molecule X undergoing a gas-kinetic collision with a surface is actually taken up onto the surface. Although several recent model studies that consider dependency of γ on RH and/or T, the majority of the earlier studies use constant γ values that only vary with aerosol particle compositions (Chen et al., 2018; Evans and Jacob, 2005; Macintyre and Evans, 2010, 2011). For one study, for the uptake onto aqueous aerosols is considered with pH dependence (Thornton et al., 2008). However, another study demonstrated that the uptake is large, irrespective of the solubility in cloud water or pH (Morita et al., 2004). Therefore, we instead choose as fixed values depending on the type of particle. Indeed, from Eq. (1) it is apparent that uptake coefficients should be unimportant for uptake onto large particles such as cloud droplets. In this study, γ for cloud particles of liquid and ice phases are given based on suggestions from earlier reports (Dentener and Crutzen, 1993; Jacob, 2000). One study (Dentener and Crutzen, 1993) used a constant of 0.1 for uptake on sea salt, sulfate, and cloud particles. They also revealed that a smaller of 0.01, which had been reported as laboratory measurements, is insensitive to effects on tropospheric oxidant components. Results of another study (Jacob, 2000) indicated constants and for uptakes on both liquid clouds and aerosols, the latter aiming to involve HO2 scavenging by clouds without accounting for details of aqueous-phase chemistry. For ice crystals, Jacob (2000) suggested based on a report by Cooper and Abbatt (1996). Jacob (2000) recommended using for hydroxy-RO2 group produced by oxidation of unsaturated hydrocarbons and for PA. The γ values for aerosols are assumed to be fundamentally the same as those for liquid cloud particles in this study. It is noteworthy that the γ values for cloud particles are given tentatively in this study and are adjusted based on evaluation of the resulting species concentrations of O3, NOy, and OH with the observations.
2.3 Experiment setup
In this study, simulations of two types were conducted to isolate the distinct effects of each HR for the surface types considered in the model (Tables 3 and S1). Whereas a control simulation standard (STD) run considers all HRs, cases with no HRs (noHR) cases intentionally ignore one or all of the HRs to calculate effects of individual HRs. The sensitivity runs turned off the separate HRs onto clouds (liquid and ice), and aerosols were also added to exploit the separate aerosol-heterogeneous and cloud-heterogeneous effects, as suggested in many earlier studies (Apodaca et al., 2008; Jacob, 2000; Lelieveld and Crutzen, 1990, 1991; Morita et al., 2004). All simulations were run in the 2009–2017 timeframe, with 2009 being treated as a spin-up year. The HR effects are determined as the differences between noHR cases and an STD simulation as in Eq. (5):
where STDi stands for the concentration of investigated atmospheric component i in the STD run and noHR(j)i denotes the concentration of component i in the sensitivity run in which the HRs of/onto j was ignored (j could be N2O5, HO2, RO2, clouds, aerosols).
An additional sensitivity test was run to examine the sensitivity of the troposphere's responses with the amplified HRs magnitudes (Table S1). These simulations only apply for HR(N2O5) and HR(HO2) to verify some uncertainties that have been argued among earlier studies (Chen et al., 2018; Evans and Jacob, 2005; Macintyre and Evans, 2010, 2011).
Table 1Computation packages in the chemistry–climate model CHASER.
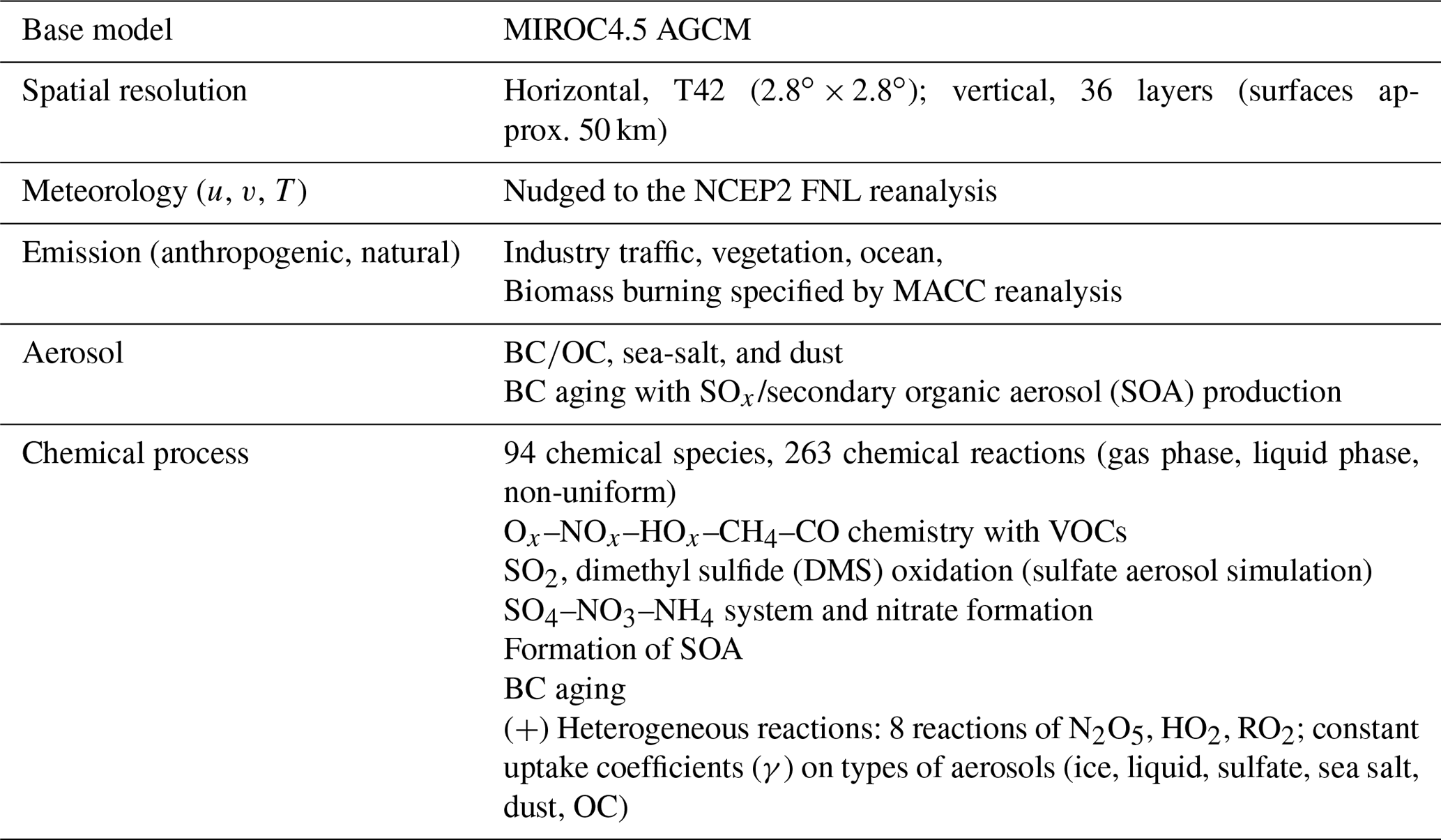
Table 2Heterogeneous reactions in CHASER.
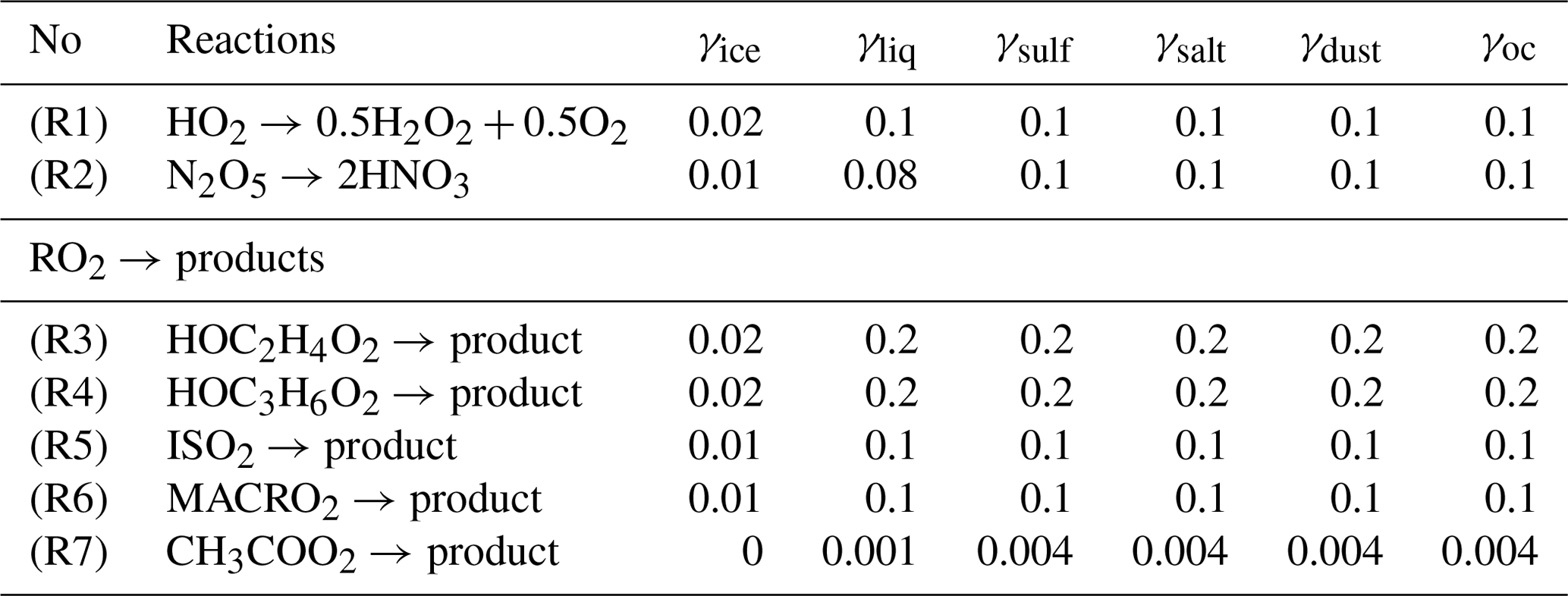
References and the details of these adjustments are given in the main text. The RO2 uptakes are assumed with inert products, as suggested by Jacob (2000). ISO2 is denoted for peroxy radicals from C5H8+ OH, and MACRO2 stands for peroxy radicals from methacrolein (CH2= C(CH3)CHO).

2.4 Observation data for model evaluation
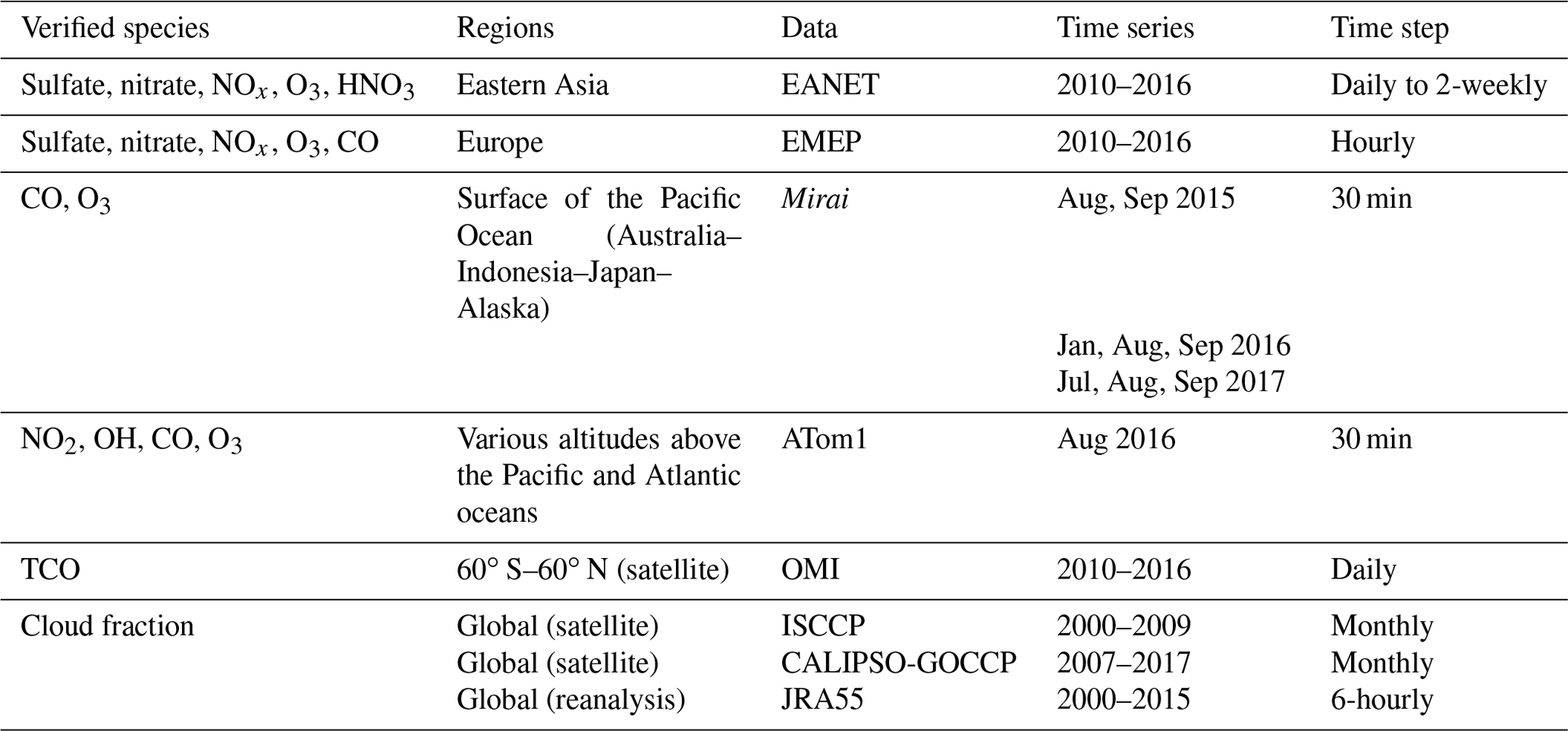
Model simulations with and without HRs are evaluated distinctively with stationary, ship-based, aircraft-based, and satellite-based measurements. The observational information, locations of the surface sites, and ship or aircraft tracks for the observations used for this study are summarized in Table 4 and Fig. 1.
EANET is well known as the Acid Deposition Monitoring Network in eastern Asia. The monthly data from 45 stations over 13 countries during 2010–2016 were used to verify surface concentrations of aerosols (sulfate, nitrate) and trace gases (HNO3, NOx, O3) in eastern Asia. We also used data of the European Monitoring and Evaluation Programme (EMEP), which compiles observations over 245 European stations.
Additionally, we exploited ship-based observational data from R/V Mirai cruise (http://www.godac.jamstec.go.jp/darwin/e, last access: 30 June 2020) undertaken by the Japan Agency for Marine-Earth Science and Technology (JAMSTEC). This study used data for surface CO and O3 concentrations in summer 2015–2017 along the Japan–Alaska and Japan–Indonesia–Australia routes (Kanaya et al., 2019). The model data were compiled in hourly time steps and were interpolated corresponding with the Mirai time step and coordinates. For verification of the vertical tropospheric profiles, we used Atmospheric Tomography (ATom1) aircraft measurements (https://espo.nasa.gov/atom/content/ATom, last access: 30 June 2020) for NO2, OH, CO, and O3.
The simulated tropospheric ozone was also evaluated using the tropospheric column O3 (TCO) derived from the OMI satellite data (https://daac.gsfc.nasa.gov/, last access: 25 February 2020). For distribution of the cloud fraction, satellite data from International Satellite Cloud Climatology Project (ISCCP, https://isccp.giss.nasa.gov/, last access: 12 June 2020), GCM-Oriented CALIPSO Cloud Products (CALIPSO-GOCCP, https://eosweb.larc.nasa.gov/project/calipso/calipso_table, last access: 12 June 2020), and Japanese 55-year reanalysis (JRA-55 – https://doi.org/10.5065/D6HH6H41, Japan Meteorological Agency/Japan, 2013) were used.
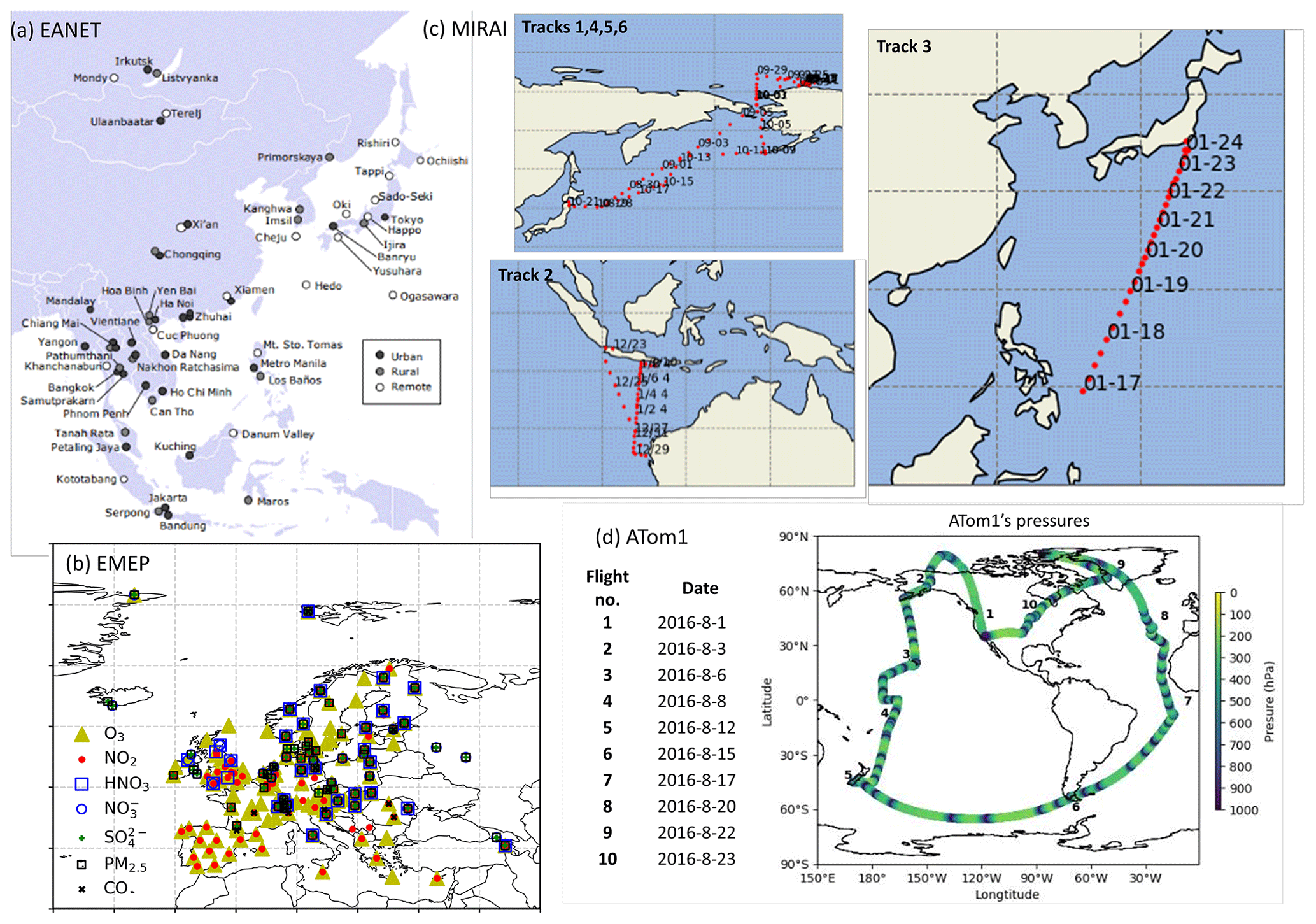
Figure 1Locations of EANET stations (a), EMEP stations (b), Mirai cruises (c), and ATom1 flights (d). The source for panel (a) is https://monitoring.eanet.asia/document/overview.pdf (last access: 12 June 2020).
Model bias and normalized root-mean-squared error (NRMSE) for each species were calculated as shown below, where n is the number of available data (number of stations × time step).
3.1 Model verifications
3.1.1 Cloud verification
For this study, we tentatively consider HRs on the cloud particle surface. Given the great uncertainties related to the reaction coefficient (γ) (Macintyre and Evans, 2010, 2011), the cloud distributions must be examined adequately in the model to the greatest extent possible. The model-calculated cloud distributions were verified using satellite observation data ISCCP D2, CALIPSO-GOCCP, and reanalysis data JRA55.
For the entire troposphere, the calculated cloud fraction was generally underestimated against the satellite observations and reanalysis data (Fig. 2, the first row). In the North Pacific region in JJA (Fig. 2, the second row), when the cloud fraction peaked in the region, the model was able to reproduce the satellite observations (ISCCP and CALIPSO). However, for the lower troposphere over the region, the cloud fraction calculated using CHASER in JJA appears to be overestimated (Fig. 2, the fourth row), suggesting that the resulting HR effects would also be exaggerated to some extent.
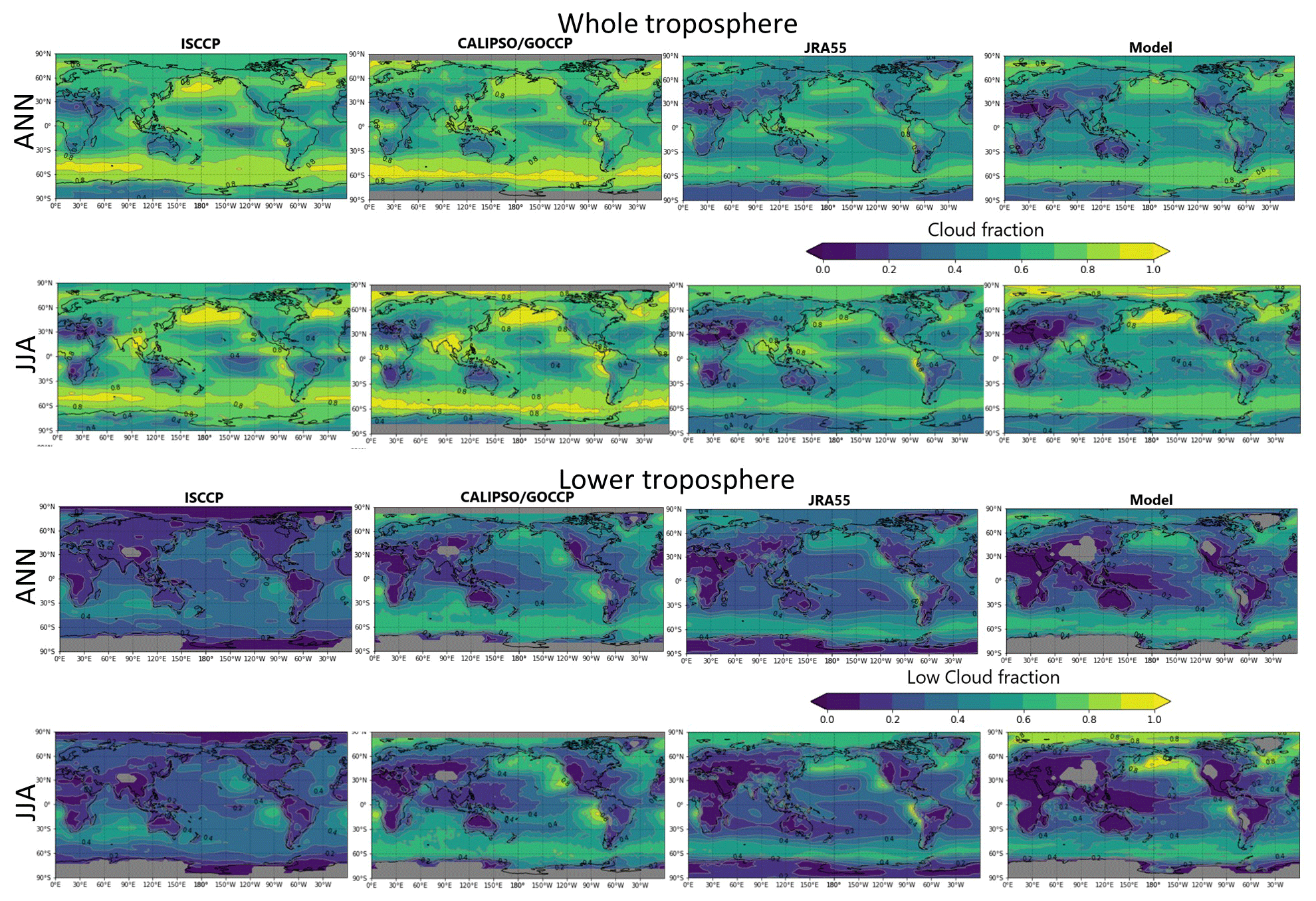
Figure 2Comparisons for cloud fraction in the whole troposphere (first and second rows) and lower troposphere (third and fourth rows). ANN denotes annual mean, and JJA denotes June + July + August mean. The first column is for ISCCP (2000–2009), the second column is for CALIPSO/GOCCP (2007–2017), and third and fourth columns are for JRA55 and CHASER (2000–2015), respectively. Color bars are the same for all panels. In ISCCP and CALIPSO data, the pressure boundary layer of the low troposphere is >680 hPa. In JRA55, the low troposphere was defined as 850–1100 hPa of pressure.
3.1.2 Verification with stationary observations
Verifications with EANET and EMEP stationary observations were conducted to assess the model performance on land domains of eastern Asia and Europe, particularly addressing the roles of the heterogeneous reactions considered for this study.
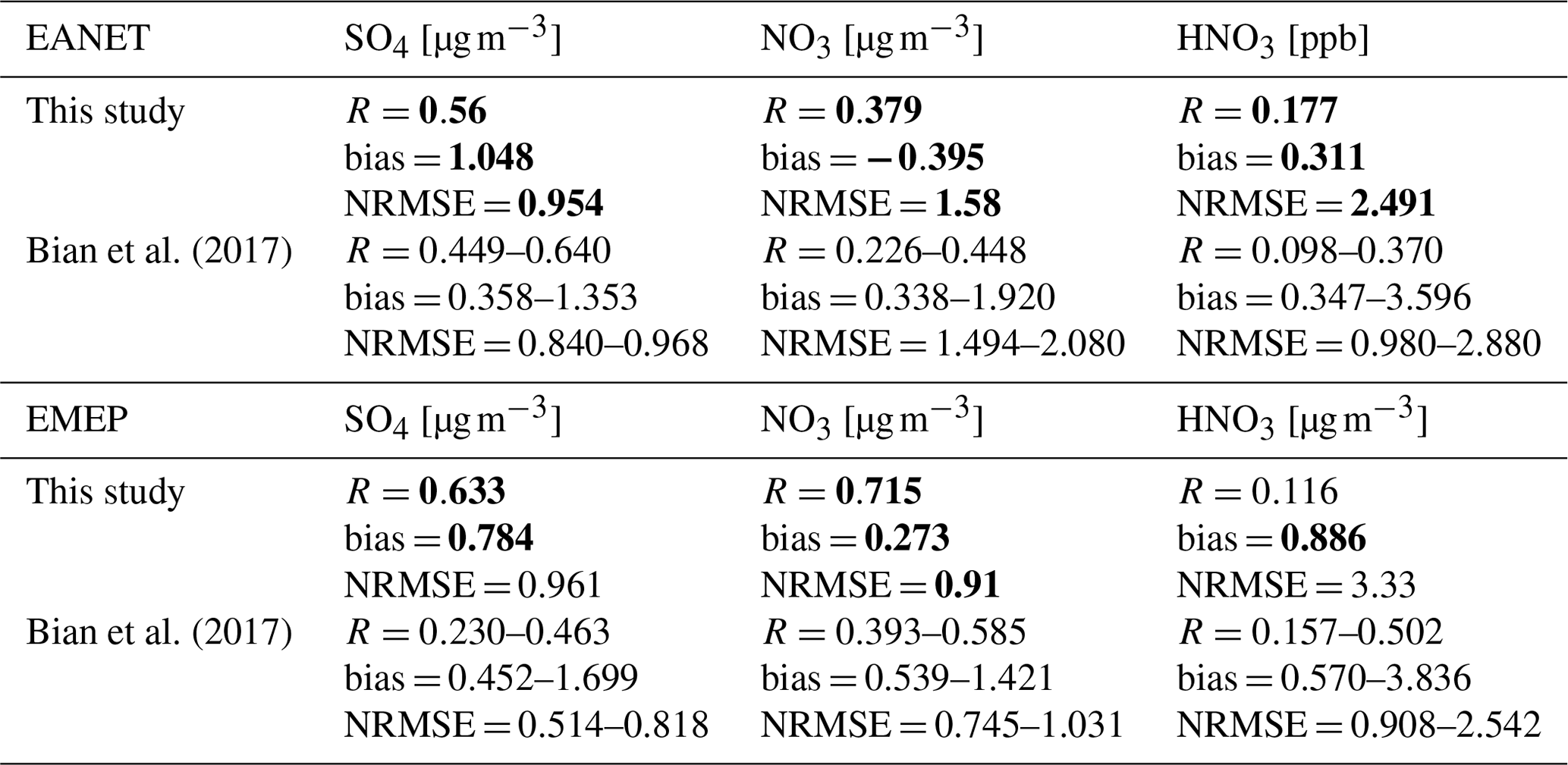
The mass concentrations of particulate matter (PM2.5); sulfate (SO); nitrate (NO); and gaseous HNO3, NOx, O3, and CO (CO only for EMEP) of 2010–2016 were evaluated (see Figs. S1 to S8 for monthly concentrations and Fig. S9 for correlations). In general, the model can moderately reproduce the PM2.5, SO, and NO aerosol concentrations at these locations (R=0.3–0.7, Table 5), although PM2.5 was underestimated, sulfate was overestimated slightly. Nitrate was underestimated for EANET and overestimated for EMEP. It is noteworthy that the model performance for EMEP stations was better than that for EANET. The PM2.5 concentration was better estimated with the inclusion of N2O5 and HO2 uptakes (bias reduction in Table 5). Figure 3a–g present the median values of NOx, O3, and CO for grouped stations in the Chinese and South Korean regions (stations in China: Jinyunshan; stations in South Korea: Kanghwa, Imsil, Cheju), remote stations with low NOx levels of EANET, and all EMEP stations. Figure 3h–n show changes in NOx, O3, and CO for these stations. The model's positive bias for NO at Kanghwa as a remote area is different from the model underestimates at other EANET stations (e.g., Bangkok, Hanoi, and Hongwen in Fig. S1). These high negative biases for NO can be associated with undervaluation for NOx and can thereby lessen the effects of N2O5 uptake.
Nitric acid in both regions was overestimated. The correlations, biases, and normalized root-mean-square error (NRMSE) of the model for SO, NO, and HNO3 are in the ranges reported in a multi-model study by Bian et al. (2017) (Table 6).
The NOx concentration for eastern Asia and Europe was underestimated, with significant bias for polluted Asian locations (Bangkok, Metro Manila, Nai Muaeng, Samutprakarn, Si Phum, Ulaanbaatar, not shown). In Fig. 3a and c, simulated NOx levels still underestimated the observed values for Chinese and European regions, at which the observed NOx could reach 16 and 7 ppb, respectively. For the low-NOx EANET region, excluding the abovementioned sites (Fig. 3b), simulated NOx levels turned to overestimate the observed levels in January, February, September, and October. The increasing effects of NOx attributable to heterogeneous reactions, although minor, mitigated these underestimations (Fig. 3a–c). Although NOx was partly reduced via uptake of N2O5, the NOx level was mostly increased because of HO2 and RO2 uptakes (Fig. 3h–j).
In this comparison, the low correlations of the model with EANET and EMEP sites for HNO3 and NOx are still a problem. The high biases for nitrogen species could be ascribed to the low horizontal resolution in this study (∼2.8∘). Higher resolutions could improve the model reproduction for surface NOx as previously investigated by Sekiya et al. (2018). Moreover, the low reproducibility of the model for NOx is probably caused by lacking mechanisms that reduce HNO3 and enhance NOx in our model. One possible mechanism is the heterogeneous reaction of HNO3 on soot surfaces (Reaction R8) (Akimoto et al., 2019, and references therein): HNO3+ soot → NO + NO2 (Reaction R8).
The additional Reaction (R4) followed by NO2 uptakes onto soot (Jacob, 2000), NO2+ particles → 0.5 HONO + 0.5 HNO3 (Reaction R9), can be expected to increase NO and decrease O3 via the consequent titration reaction. These changes could reduce the model overestimates for HNO3 and O3 and the model underestimates for NOx with EANET and EMEP stations. Further tests for this issue shall be discussed in a future report.
CO for EMEP was partly underestimated by the model, especially during January–March (Fig. 3g). This underestimate was mitigated by increasing effects because of HRs of N2O5 and HO2. The uptakes of RO2, in contrast, minorly reduced CO levels (Fig. 3n) so that the model bias was worsened slightly. For O3, whereas the model tends to overestimate this tracer for both regions (Fig. 3d–f), O3 reduction effects of all HRs (Fig. 3k–m) alleviated the model overestimates from April to December, although advanced reduction is still needed. In January–March, the model tended to underestimate O3 levels (Fig. 3d–f), which was exaggerated by reduction effects for O3. In general, the STD simulation with coupled HRs partly improved the agreement related to the particulate and gaseous species, showing less bias than that of simulations without HRs (Table 5).
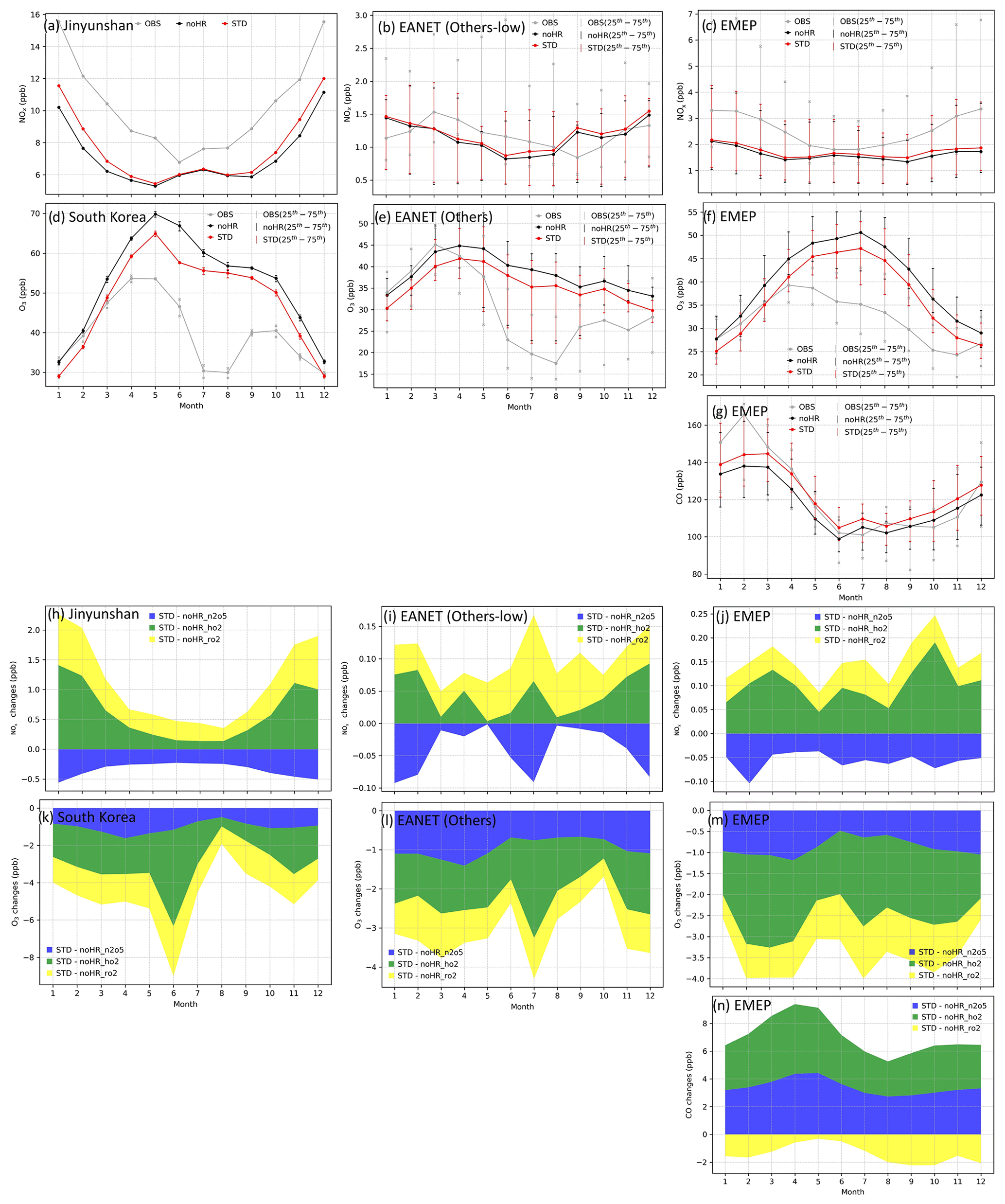
Figure 3Monthly averaged concentrations at EANET and EMEP from 2010–2016 (a–g) and corresponding HR effects (h–n) for NOx: (a, h) Jinyunshan (China), (b, i) other low-NOx EANET stations, and (c, j) EMEP stations; for O3: (d, k) Korean stations, (e, l) other EANET stations, and (f, m) EMEP stations; for CO: (g, n) EMEP stations. In (a)–(g), grey lines are observations (OBS), red lines are the STD simulation, and black lines are the noHR simulation. All lines showed the median of monthly mean concentrations for each group of stations, except (a). In (b)–(g), vertical thin lines with markers show the 25th–75th percentiles of monthly mean concentration at the particular group of stations. In (h)–(n), blue fields are changes caused by HR(N2O5), green fields are changes in HR(HO2), and yellow fields are changes caused by HR(RO2).
Table 5Model correlations and biases with EANET/EMEP observations: three-sigma-rule outlier detection is applied for each station before calculating all data. For NOx, all data were filtered once more using the two-sigma-rule. Bias of the sensitivity run is shown in bold if it is higher than the bias of the STD run. R has no unit; the units in brackets are for biases.
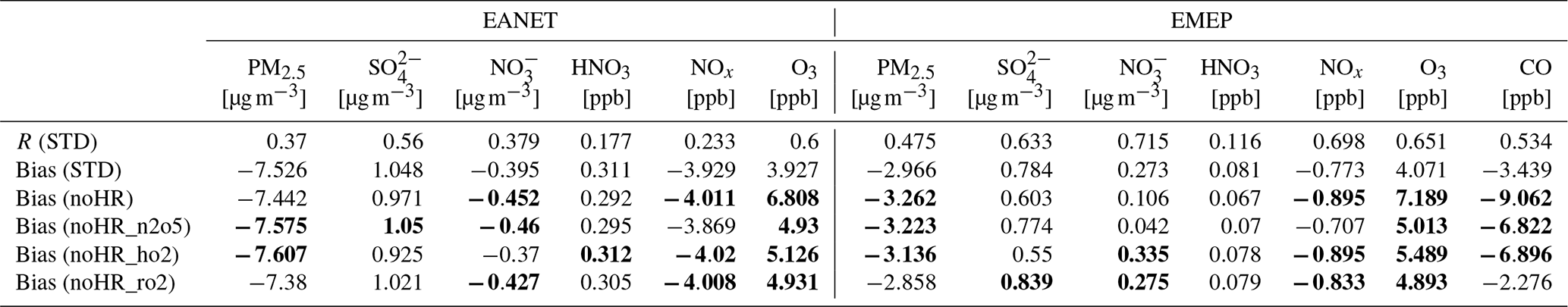
Table 6Comparisons between EANET and EMEP observations with atmospheric models. Outlier detection follows that in Table 5. The model result is shown in bold if it is better than or agreed with Bian's report. R has no unit; units in brackets are for biases and NRMSE.
3.1.3 Verification with ship-based measurements
The model simulations were also verified with O3 and CO observations from the Research Vessel (R/V) Mirai for the Pacific Ocean region. This study specifically examines data from the four cruises of R/V Mirai for the Japan–Alaska region (40–75∘ N, 140∘ E–150∘ W) in summer, designated as MR15-03 leg 1 and leg 2 (28 August–21 October 2015, labeled as Track 1 in this study), MR16-06 (22 August–3 October 2016 as Track 4), MR1704 leg 1 (11 July–2 August 2017 as Track 5), and MR1705C (24 August–29 September 2017 as Track 6). Two other cruises during DJF for the Indonesia–Australia region (5–25∘ S, 105–115∘ E) and Indonesia–Japan region (10–35∘ N, 129–140∘ E) are also explored in this study, respectively designated as MR15-05 leg 1 (23 December 2015–10 January 2016 as Track 2) and MR15-05 leg 2 (17–24 January 2016 as Track 3). All measuring data for CO and O3 from the six cruises are respectively plotted in Fig. 4a and b as grey dots. The ship-based data used in this study was partly reported (T1–4) in the work of Kanaya et al. (2019), including the extraordinary peak of CO on 26 September 2016 exceeded 500 ppbv (off the scale in Fig. 4a-T4) associated with heavy fires in Russia (Kanaya et al., 2019). Data for the North Pacific region (40–60∘ N) are addressed in light-blue shades in Fig. 4 (T1, T4–6) for analysis in Sect. 3.2.
Table 7 shows correlation coefficients (plotted in Fig. S10), indicating that the CHASER simulations for CO and O3 are in good agreement with Mirai observations (R= approx. 0.6). However, the model still shows some discrepancies for both CO and O3 concentrations. In general, the estimated CO and O3 are both reduced for T1 and T4–6 as compared to observations, whereas they are superior for the data located in 20∘ S–20∘ N during T2–3. Overestimations for CO and O3 occurring in the region with considerably low levels of these species might be attributed to the lack of halogen chemistry in the model, as also discussed for the nearby region in a past report (Kanaya et al., 2019). Underestimates for O3 levels up to 70 ppbv in the higher latitudes (Fig. 4b: T1, T4–6) are ascribable to the insufficient downward mixing process of stratosphere O3 in the model (Kanaya et al., 2019). Except for the CO's peak on 26 September 2016 mentioned above, the mild reductions for CO (<30 ppbv) in the model during T1 and T4–6 as compared to the observations could be attributed to the insufficient emission from territories, international shipping, and aviation, as we used the HTAP-II emission inventory. These reductions for CO in Kanaya's work are minor, e.g., for T1 (MR15-03 leg 1 and 2 in their work), due to reanalysis data with finer horizontal resolution (1.1∘) utilized for the CO emission rate (Kanaya et al., 2019).
The negative biases in the noHR simulations for CO are lower in the STD run for all cruises, as they are for the North Pacific region (second versus third/fourth/fifth data rows for CO, Table 7). The CO-increasing effects by N2O5 and HO2 uptakes (Fig. 4c) are consistent with the comparison for EMEP. This is also true for CO-reduction effects because of HR(RO2). Whereas the effects by N2O5 and HO2 reduce the model bias, the CO-reducing effects by HR(RO2) exaggerated the CO bias (second versus sixth data rows for CO in Table 7), which is already apparent for comparison with EMEP (last column, Table 5).
For O3 level, the model underestimates (Table 7) are in the opposite direction to the O3 overestimates for EANET and EMEP stations (Table 5). The lower panels presented in Fig. 4b show marked O3 reduction with all HRs (gaps between red and black lines), mostly contributed from the HO2 uptake onto cloud particles (Fig. 4d: green and hatched fields). This marked reduction of the O3 level is evident at some points during the cruises, especially in the North Pacific region (the shaded areas) and for T5. Unlike comparisons for land domain data (Table 5), O3 reduction because of HRs worsens the model underestimates during the Mirai cruises. It is noteworthy that one cannot necessarily confirm whether the STD run simulates these species better than the noHR does because tropospheric CO and O3 levels are controlled by a complicated chemical mechanism and an interplay of emission, transport, deposition, and local mixing in the boundary layers. As discussed later in Sect. 3.2, the surface aerosols concentration in the western Pacific Ocean is mostly dominated by liquid clouds (exceeding 50 000 µm2 cm−3 during JJA) and sulfate aerosols (approximately 75 µm2 cm−3 in JJA). The model improvements in reproducing CO by adding N2O5 and HO2 uptake indicate that the appropriate mechanisms of these processes onto cloud droplets and sulfate aerosols are well established in the model. For HR(RO2), which induces the smallest and opposite effects on CO compared with the effects of N2O5 and HO2 uptakes, it can be stated in general for the total HR effects that including all three HRs partially improves the model during Mirai cruises.
Table 7Model correlations and biases for Mirai. No outlier filtration is applied. The bias of the sensitivity run is shown in bold if it is higher than the bias of STD run. R has no unit; the units in brackets are for biases.
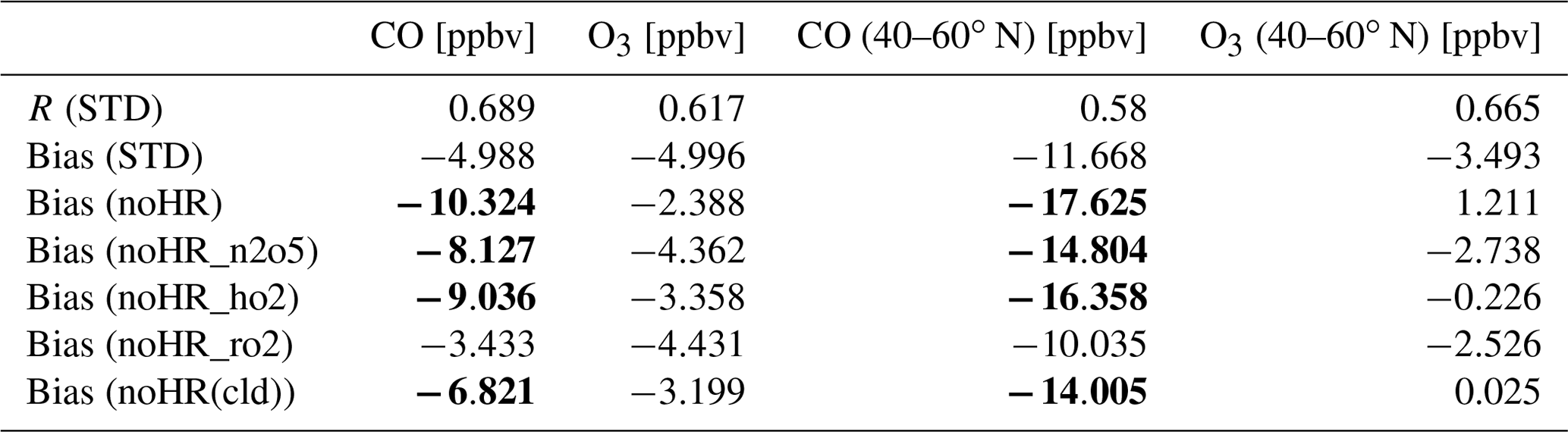

Figure 4Observed and simulated concentrations for CO (a) and O3 (b) and their changes caused by each HR (c, d) during Mirai cruises. The left axis shows concentrations. Dashed lines show latitudes of cruises scaled with the right axis. The horizontal axis shows cruise travel times. Light-blue areas show data in the North Pacific region (140–240∘ E, 40–60∘ N). In (a) and (b), grey dots are for observations, red lines are for STD simulations, and black lines are for noHR simulations. In (c) and (d), blue fields are for changes in HR(N2O5), green fields for changes in HR(HO2), yellow fields for changes in HR(RO2), and hatched fields for changes in HRs (cloud). Note that in (d) all three fields are stacked but in (c) only blue and green fields are stacked for better illustration of negative and positive changes.
3.1.4 Verification using aircraft measurements
To verify the model performance in the free troposphere, we used ATom1 aircraft measurements in August 2016 (for NO2, OH, CO, and O3). Table 8 lists the model's correlation coefficients and biases of each sensitivity run against ATom1. Figure 5 shows observed and simulated concentrations of CO and O3 during flight no. 2 in the North Pacific (NP) region. Figure 6 exhibits vertical biases of the model and computed HR effects caused by each HR for all ATom1 flights and NP region. The spatial and temporal concentrations are available in Fig. S11. Correlations are shown in Fig. S12.
In general, the model simulations for NO2, OH, CO, and O3 adequately agree with aircraft measurements with R>0.5 (Fig. S12). However, NO2 and CO still tend to be underestimated by the model, which is consistent with comparisons for EANET/EMEP and Mirai observations. In Fig. 5, the CO-increasing effects, mostly due to the uptake of N2O5 and HO2 (Fig. 6o), mitigated the negative bias of the model. This CO bias reduction was visible for all flight altitudes, the lower troposphere, and the North Pacific region (Table 8; Fig. 6d and h). Both N2O5 and HO2 uptakes show improvements for CO reproduction of the model. However, RO2 uptake seems to worsen the model's CO bias due to its reducing effect for CO (Fig. 6o and s), which is consistent with the Mirai comparison.
For the O3 level, the model generally overestimates O3 when calculating for all altitudes or lower troposphere, which is similar to the EANET/EMEP observations. In the North Pacific region with P>600 hPa (40–60∘ N, 198–210∘ W), the model bias for O3 in STD run turns to underestimate (second data row and second column from the right, Table 8), which might be similar with Mirai data verification for the western North Pacific (143∘ E–193∘ W). However, the underlayers (>700 hPa) again show overestimation (second data row – last column, Table 8). As Mirai and ATom1 data show, the underestimates for O3 exist at the marine boundary layer in the western North Pacific and extend to the upper troposphere (<700 hPa) of the east side, which might be ascribed to the insufficient downward mixing process of stratosphere O3 in the model as discussed previously.
The HR effects on O3 are generally negative effects (Fig. 6m), although they are small and barely recognizable in Fig. 5, which mitigates the model bias in the noHR run. This model improvement is consistent for all flight altitudes, the low troposphere, and the North Pacific region (second versus third data rows in Table 8). Both HR(N2O5) and HR(RO2) typically contribute to this improvement (Fig. 6m and q). In contrast, HR(HO2) seems to only reduce the model bias in a thin layer: from the ground up to 800 hPa for all flights and 700 hPa for the North Pacific region (Fig. 6m and q). At the bottommost layers in this region, the model's overestimates for O3 are reduced by the negative effects of HO2 uptake (Fig. 6f and q). The extension of model bias because of HO2 uptake above 800 hPa is attributable to its increasing effect on O3 level (Fig. 6m). We recognize that this O3 increase effect above 800 hPa is opposite to the effects for EANET/EMEP and Mirai comparisons, which is discussed in Sect. 3.2 for HO2 uptake effects.
The vertical means of model biases and changes for all four species (NO2, OH, CO, O3) are presented in Fig. 6. In general, the STD run reduces model biases for all four species, with better performance for broader regions (all flights) than for the smaller region (North Pacific). In the North Pacific region, the negative bias for O3 is observed only for the 500–900 hPa layers (Fig. 6f). The model bias is apparently extended in this region. However, the inclusion of HR(HO2) leading to O3 increment (Fig. 6f) reduces O3 bias in this region, which might indicate that the O3 increase effect by HR(HO2) is verified, particularly in 500–900 hPa layers during ATom1.
We also verify the total uptake of N2O5, HO2, and RO2 onto ice and liquid clouds using data obtained from ATom1 flights within the free troposphere. As Table 8 shows, the inclusion of HRs onto clouds reduces the model biases for CO and O3 in all calculations. In Fig. 6l–s, because the HRs(cloud) effects occupy the major part of total HRs effects for NO2, O3, and OH, especially for NP region and low troposphere (>800 hPa), cloud uptakes could also contribute to the overall reduction in model bias against ATom1. For O3, HRs(cloud) mostly induce negative effects (Fig. 6m and q). At the layer of 600–800 hPa in the NP region, this O3 reduction due to HR(RO2) onto clouds (Fig. 6q: yellow and hatched patterns) might account for the model worsening in O3 levels as described above. This result might prove that cloud overestimation for the North Pacific, as revealed at the beginning of this section, affects the model bias in this region.
Table 8Model correlations and biases with ATom1: three-sigma-rule was applied for CO and O3. NP denotes North Pacific region (140–240∘ E, 40–60∘ N). The bias of the sensitivity run is presented in bold when it is higher than the bias of STD run. R has no unit; the units in brackets are for biases.
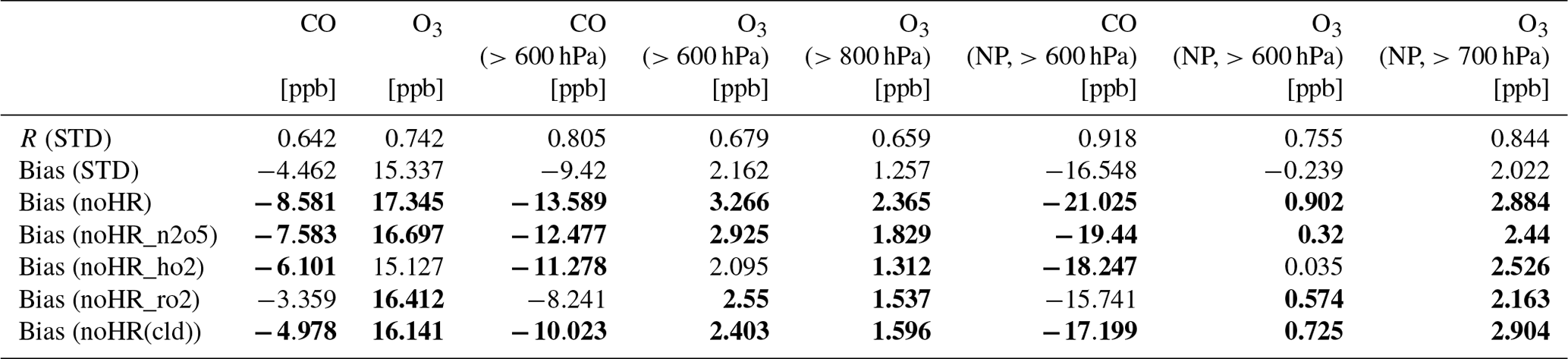

Figure 5Observations and simulations for CO and O3 during ATom1 flight 2 (198–210∘ E, 20–62∘ N). Blue shaded areas show data for P>600 hPa.
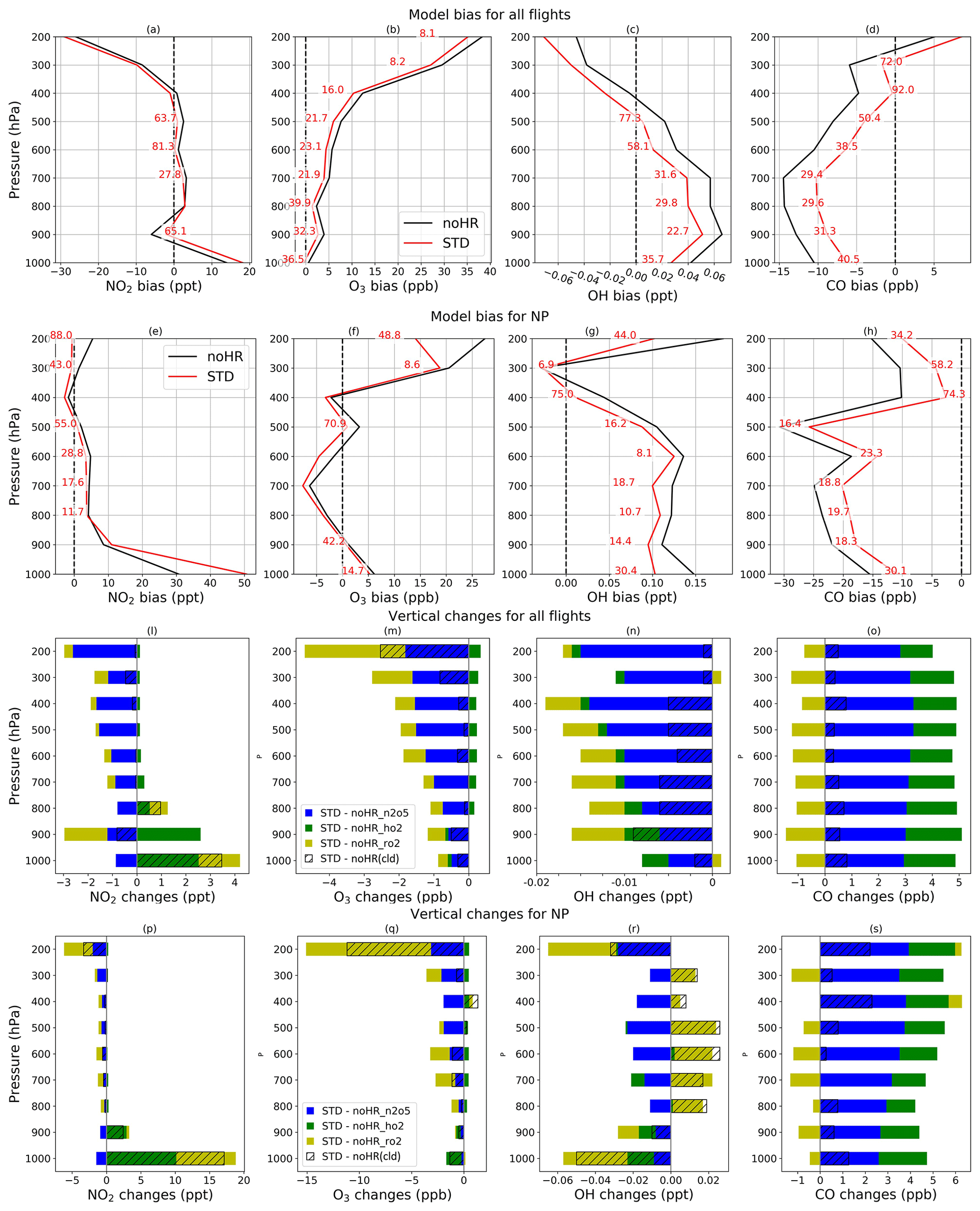
Figure 6Vertical bias against ATom1 (a–h) and vertical HR effects (l–s). Data for each pressure level P are calculated within the range of P±50 hPa, with the applied three-sigma-rule for outlier detection. All rows show calculations for all flight domains the and the North Pacific region. The horizontal axis shows model bias and absolute changes with units written in each panel. The vertical axis shows pressure (hPa). The red numbers in (a)–(h) represent relative reductions (%) of the STD run's bias compared to that of the noHR run.
3.1.5 Verification with OMI satellite observation for TCO
We also tested STD and noHR simulations using the tropospheric column ozone (TCO) derived from the OMI satellite instrument (Figs. S13 and 7). In a large area of the Northern Hemisphere, the inclusion of HRs (STD run) generally improved the consistency with the OMI TCO, as seen in Fig. 7b, d, f with less overestimation than Fig. 7a, c, e, particularly enhancing the winter minima (first and second panels in Fig. S13). This improvement in DJF is attributed mostly to the reductive effects of HR(N2O5) and HR(RO2) in the lower (800 hPa) and middle troposphere (500 hPa), respectively (see Fig. 10 for vertical profiles of HR(N2O5) on O3 and Fig. 13 for vertical profiles of HR(RO2)). In the North Pacific, HRs appeared to exaggerate O3 underestimates, especially for latitudes higher than 40∘ N (Fig. 7) during the first half of the year (third panel, Fig. S13). However, such a discrepancy, which was also observed from comparison for R/V Mirai observations (Fig. 4), might result from other factors such as deposition or vertical mixing rather than by HRs.
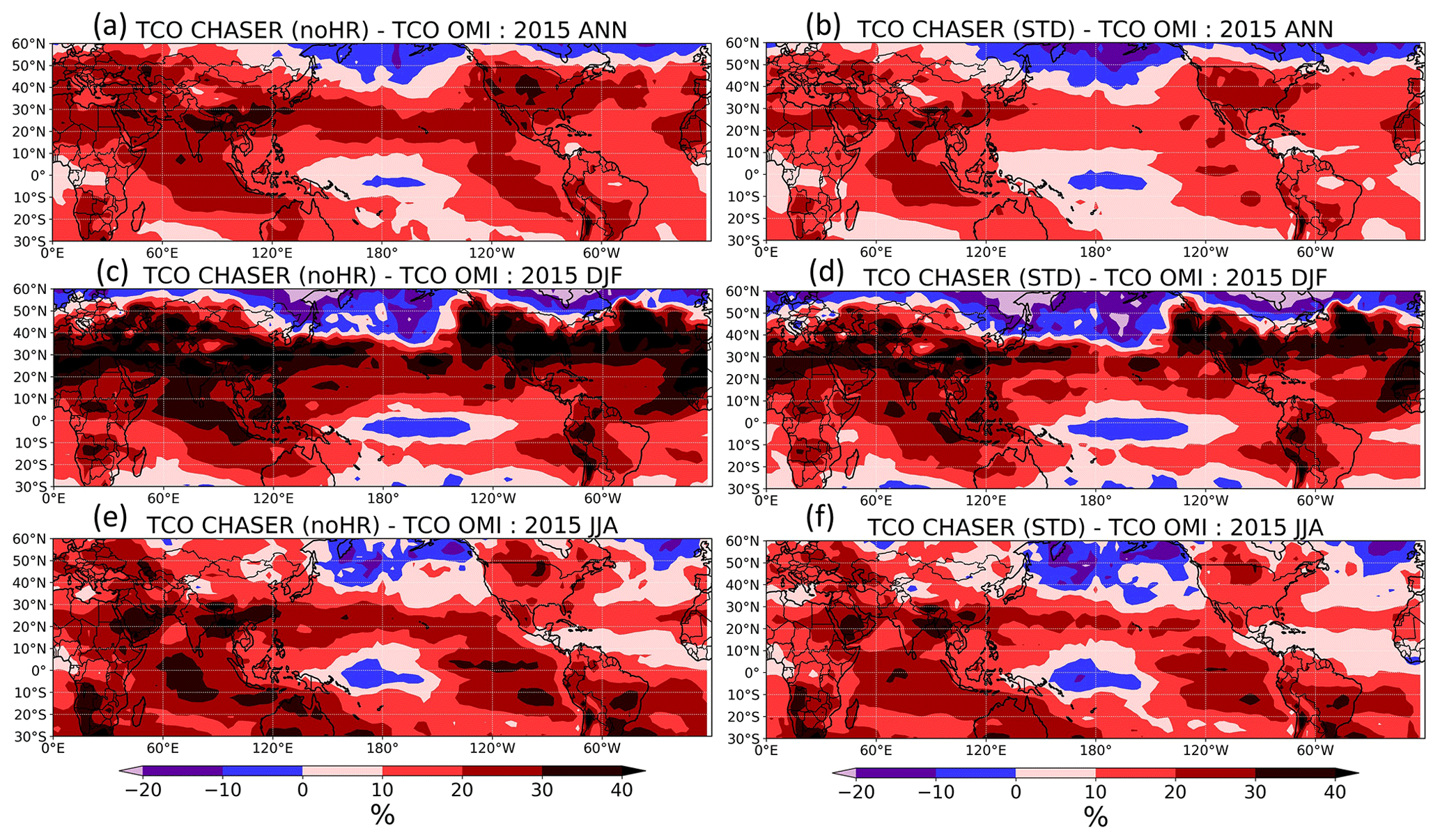
Figure 7Differences in tropospheric column ozone (TCO) in CHASER's noHR (a, c, e) and STD (b, d, f) runs from OMI: ANN is annual; DJF is December, January, February; and JJA is June, July, and August.
3.2 HR effects
This section presents a discussion of the global effects of HRs calculated using CHASER with their spatial distributions in the troposphere using standard (STD) and sensitivity simulations (noHR_n2o5, noHR_ho2, noHRs_ro2, noHR) for the meteorological year of 2011. Aside from the main simulations described in Table 3, additional runs that separately turned off the uptakes onto clouds or aerosols for each HR are also conducted to exploit the contributions of effects to the troposphere.
3.2.1 Distribution of clouds and aerosols surface area density (SAD)
To obtain the parameters for uptake to clouds and aerosols, SAD estimations are used together with cloud fraction and aerosols concentration. Hereinafter, we discuss SAD distributions for total aerosol, ice clouds, and cloud droplets, which are estimated for the model using Eqs. (2), (3), and (4), respectively.
In Fig. 8, total surface area concentrations of liquid clouds and aerosols are both much lower aloft than at the surface (as counted on the dry and wet depositions). The liquid cloud SAD results are 2 orders of magnitude larger than ice cloud SAD and total aerosol SAD. The ice cloud SAD, distributed at the middle and upper troposphere, is enhanced for N/S middle latitudes in wintertime. Liquid cloud SAD concentrates mainly at the surface with distributions extending to 500 hPa and maximized at approx. 800 hPa over the mid-latitude storm tracks and in tropical convective systems, especially at 60∘ N in JJA. Total aerosol SAD was derived mainly from pollution sources at 40∘ N during both seasons, with higher concentrations apparent for DJF and a greater spatial spread observed for JJA. Sulfate aerosols are becoming the dominant source of aerosol surface area in the model above 600 hPa (approx. 20 µm2 cm−3) in addition to organic carbons and soil dust (both are approx. 10 µm2 cm−3 in JJA) for the Northern Hemisphere.
In Fig. S14, showing the SAD distribution at the surface, the SAD for liquid clouds is dominant in JJA, reaching approx. 50 000 µm2 cm−3 for mid-latitude and high-latitude ocean regions. Liquid clouds make the greatest contribution to the SAD at the surface. Our model performance for aerosol SAD shows agreement with that presented in an earlier report (Thornton et al., 2008). Sulfate aerosols are prevalent in the northern mid-latitudes near industrial bases; maximize at the surface in DJF for the Chinese region (exceeding 1000 µm2 cm−3), NE U.S. (approx. 500 µm2 cm−3), and western Europe; and transport to the North Pacific region in JJA (approx. 250 µm2 cm−3). Soil dust aerosol SAD dominate in the regions of the Sahara and Gobi deserts, reaching annual average values exceeding 100 µm2 cm−3. Organic carbon (OC) is a dominant source of aerosol SAD over biomass burning regions in China (up to 1000 µm2 cm−3 in DJF), South Africa (up to 800 µm2 cm−3 in JJA), western Europe, and South America. The black carbon (BC) surface area can reach values exceeding 600 µm2 cm−3 in DJF for the region of China or other significant industrial areas (India, which reaches 75 µm2 cm−3, NE U.S., and Europe) or over tropical forests, primarily in Africa. Sea salt aerosols are most important in high-latitude oceans during winter. However, the maximum contributions only reach 2 µm2 cm−3 in our model, which is greatly underestimated compared to Thornton's work (75 µm2 cm−3) (Thornton et al., 2008). In brief, SAD for aerosols of all types contributes the most during DJF, whereas during JJA, the SAD for liquid clouds and sulfate aerosols are dominant, particularly for the northern high-latitude and mid-latitude oceans. The total aerosol SAD in this region is approx. 75 µm2 cm−3, which is consistent with the estimation by Thornton et al. (2008).
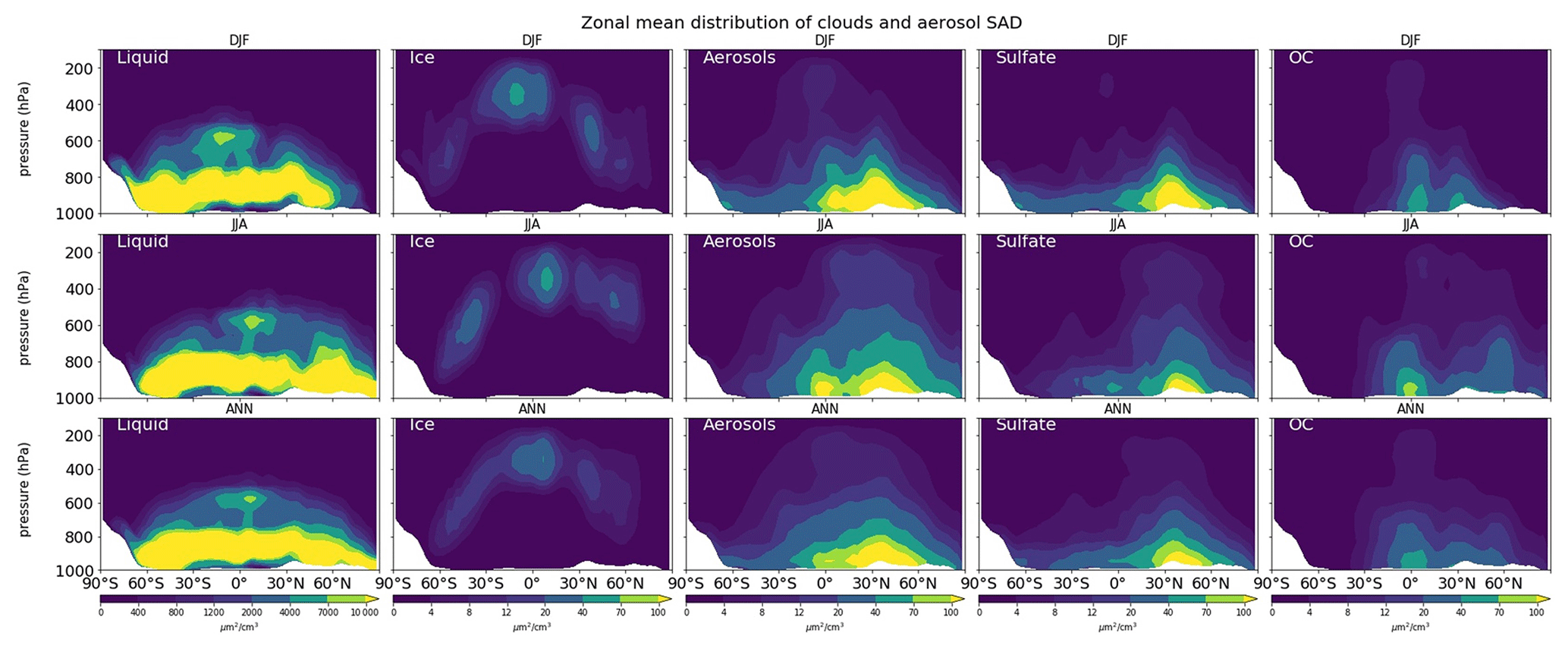
Figure 8Zonal, seasonal mean (upper and middle), and annual mean (lower) distribution of cloud droplet surface aerosol density (SAD), cloud ice SAD, total aerosol SAD, sulfate aerosol SAD, and organic carbon SAD (from left to right).
3.2.2 Effects of N2O5 heterogeneous reaction (HR(N2O5))
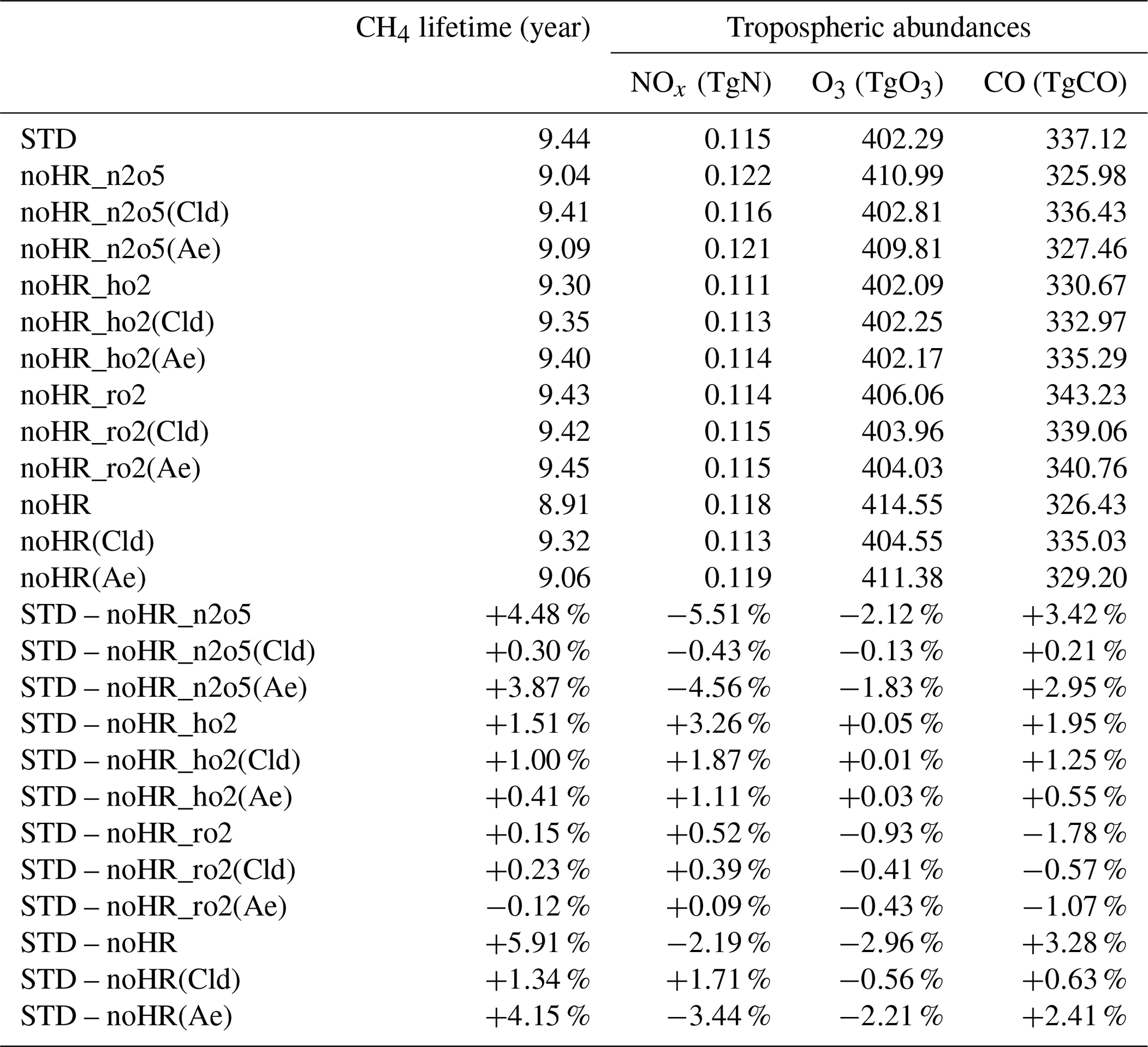
The inclusion of HR(N2O5) in the model increases global methane lifetime by +4.48 % and changes NOx, O3, and CO abundances by −5.51 %, −2.12 %, and +3.42 %, respectively (Table 9).
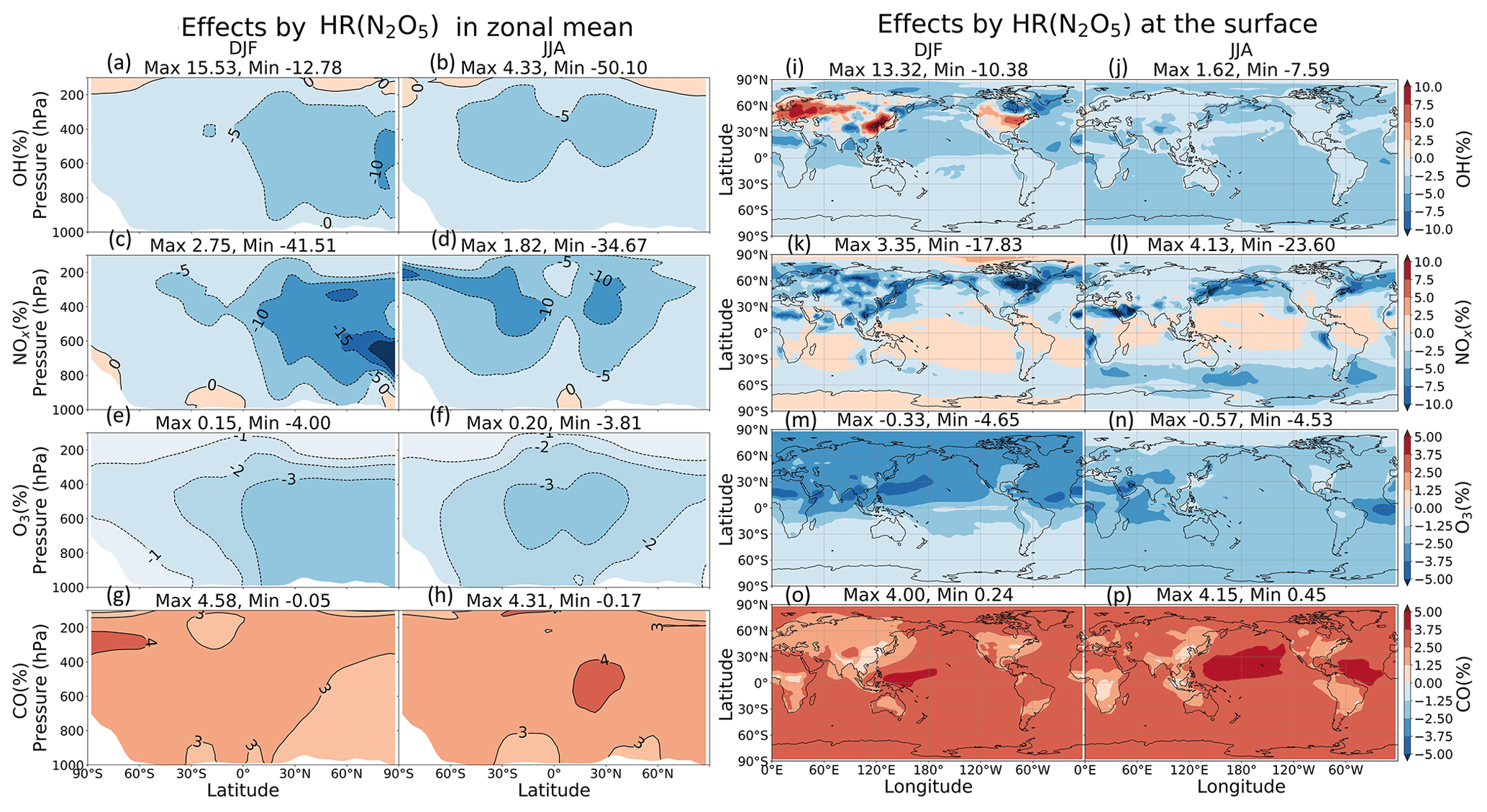
Figure 9Effects by N2O5 uptake onto both clouds and aerosols in zonal mean (a–h) and at the surface (i–p). Note that the color scale for (a)–(h) is different from that for (i)–(p).
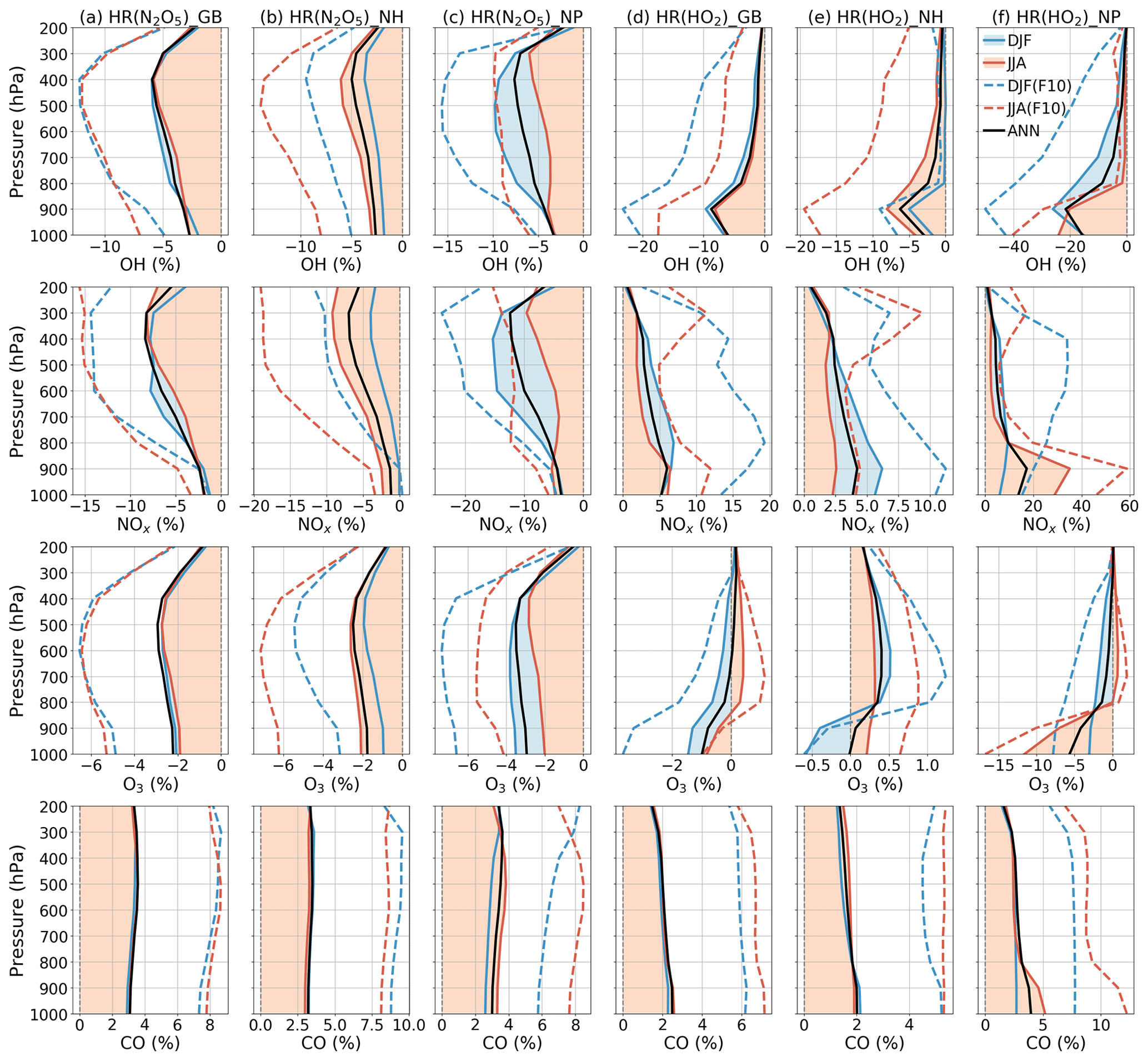
Figure 10Averaged effects of N2O5 (a–c) and HO2 uptakes (d–f) for each air pressure range. Calculations are for global data (a, d), Northern Hemisphere data (b, e), and North Pacific region data (c, f). Dashed lines show effects of the FCTHR_10 run (Sect. 3.3). The legend is in panel (f) for December–January–February (DJF), June–July–August (JJA), and annual (ANN) data.
In Fig. 9a–h, the changes in OH, NOx, O3, and CO are most significant in the middle troposphere (400–600 hPa). These changes are attributed mostly to uptakes of N2O5 onto aerosols, rather than onto clouds. Marked negative effects on NOx concentration are apparent for DJF in the middle troposphere (600–700 hPa) of the 60∘ N and the Arctic region ( % at 700 hPa) (Fig. 9c). The effects are probably associated with high concentrations of sulfate aerosols, organic carbons, or soil dusts in the middle troposphere (see the paragraph above) and are also related to a long chemical lifetime of NOy in the middle and upper troposphere in winter. When it comes to JJA, these negative effects become significant at higher altitudes around 30∘ N/S ( % at 400 hPa). At the surface (Fig. 9i–p), HR(N2O5) causes negative effects on NOx, O3, and OH concentrations (up to about −24 %, −5 % and −8 % respectively) and positive effects on CO concentration (up to about +4 %), which are also mainly attributable to the N2O5 uptake on aerosols.
In Fig. 10, the latitude–longitude (lat–long) means of HR(N2O5) effects are calculated for each pressure range (pressure ranges are defined as in Fig. 6). The global NOx decrease is up to −9 % at 300–400 hPa. This decrease causes correspondent reductions in O3 and OH, which are calculated as about −3 % and −7 % at 400–600 hPa, respectively, for lat–long mean O3 and OH. About 4 % lat–long mean CO increment throughout the entire troposphere responds to decreased OH.
The small effects of HR(N2O5) on O3 in the lower troposphere are consistent with findings from an earlier study (Riemer et al., 2003). Reductions in O3 and NOx concentrations also agree well with the collective knowledge summarized in work reported by Brown and Stutz (2012). Despite a considerable HR(N2O5) effect calculated in the middle troposphere, its effect in the whole troposphere is apparently not as great as reported to date. Another study assessed HR(N2O5) effects on annual burdens of NOx, O3, and OH as −11 %, −5 %, and −7 %, respectively, when using a similar value (0.1) (Macintyre and Evans, 2010). Although the effects of magnitude estimated in our work (Table 9) are almost half those of this earlier study (probably because of differences in NOx emissions, estimation of SAD, and chemical mechanism), the effect tendencies are similar. A strong increase of ozone attributed to N2O5 uptake under high-NOx conditions calculated using box models was reported from an earlier study (Riemer et al., 2003), but this is only slightly apparent in our global model. Our results revealed that the HR(N2O5) effect might help clean up NOx pollutant. However, it increases the concentration of other pollutants (such as CO) because of the effects of reducing oxidizing agents in the atmosphere.
3.2.3 Effects of HO2 heterogeneous reaction (HR(HO2))
Regarding the effects of HR(HO2), the tropospheric methane lifetime increases by approx. 1.51 %. Abundances in NOx, O3, and CO change, respectively, by +3.26 %, +0.05 %, and +1.95 % (Table 9). In the entire troposphere, the influences of HR(HO2) are not as large as that of HR(N2O5).
As Fig. 11a–f shows, the zonal-mean effects of HR(HO2) on NOx, OH, and O3 are more widespread in DJF but are more concentrated at the surface in JJA because of the high level of HO2. The most substantial effects by HR(HO2) are calculated in JJA at the surface of North Pacific (140–240∘ E, 40–60∘ N) by as much as about +69 % (NOx), +7 % (CO), −70 % (OH), and −21 % (O3), which are more significant than those of HR(N2O5) at the surface. In the lower troposphere, HR(HO2) suppresses the NO oxidation (Reaction R10) and thus preserves a high ratio and generally restricts OH and O3 formations.
These effects are primarily attributable to HR(HO2) in clouds rather than to aerosols (which is opposite to N2O5 uptake). These OH and O3 reduction effects go along with past studies in which approx. 50 % OH and approx. 10 % O3 of reductions are calculated for the low troposphere of northern mid-latitude region ascribed to aqueous-phase HOx sink in clouds (Lelieveld and Crutzen, 1990, 1991). The efficient scavenge of HO2 radical by cloud droplets might associate with acid–base dissociation HO2/O and electron transfer of O to HO2 to produce H2O2 (Jacob, 2000). Furthermore, cloud droplets SAD in our model are 2 orders of magnitude higher than total aerosol SAD (Fig. 8), which also contributes to the preference of the aqueous-phase HO2 sink. Our large calculated effects for the North Pacific region are new findings from other models, which have considered only aqueous aerosols (Stadtler et al., 2018; Thornton et al., 2008) because cloud particles are dominant at remote marine areas in addition to sulfate and aqueous sea salt particles (discussed at the beginning of Sect. 3.2). The HO2 uptake onto aerosols is minor; it is observed only in DJF in the Arctic region and polluted areas (China and US), with apparent changes of up to +17 % for NOx, −40 % for OH, and −14 % for O3 at the local surface (Fig. 11i, k, m). The aerosol negative effect of HR(HO2) on surface O3 concentration is significant in the Chinese area, which might be in line with other studies of the Chinese O3 trend (Kanaya et al., 2009; Li et al., 2019; Liu and Wang, 2020; Taketani et al., 2012), which suggests that the observed recent O3 increases can be attributed in part to reduced HO2 uptake under aerosol (PM) decreases brought about by the new Chinese air pollution policy.
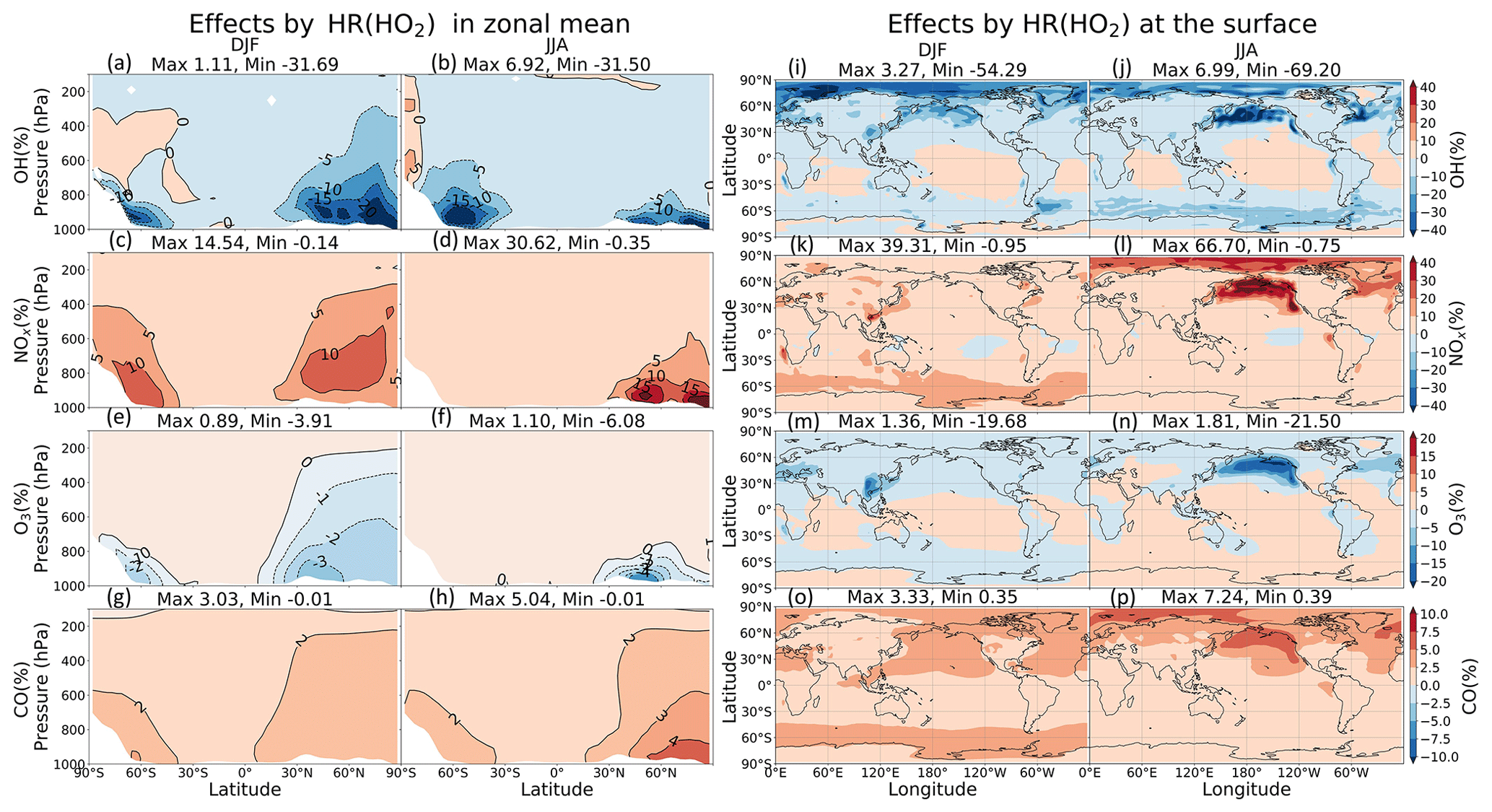
Figure 11Effects of HR(HO2) on both clouds and aerosols in zonal mean (a–h) and at the surface layer (i–p). Note that the color scale for (a–h) is different from that for (i–p).
In Fig. 10, vertical profiles show that the lat–long averaged effect of HR(HO2) on OH is −9 % in the lower troposphere. As a result, the lat–long mean CO level increases by 2 % at the surface. Additionally, the daytime NOx oxidation by OH is suppressed. Also, NOx might be preserved in clouds (Dentener, 1993), which increases the lat–long averaged NOx level by +6 % at 900 hPa. The lat–long mean O3 is reduced by −1 % at the surface, but it is increased at higher altitudes (about +0.2 % at 300 hPa). The reduction of O3 is associated with HO2 depletion in clouds and aqueous aerosols as described above, coupled with the NOx preservation in clouds, which enhances the ratio. The preserved NOx in clouds might remain available for O3 production after the cloud evaporates (Dentener, 1993), along with the low SAD for both liquid clouds and aerosols at higher altitudes (Fig. 8), thereby increasing O3 in places other than the aqueous phase. The O3 increment might be trivial in DJF but enhanced in JJA. As a result, the Northern Hemisphere mean O3 in JJA exhibits only positive effects. In contrast, for the North Pacific region in JJA, due to its large cloud fraction, an O3 reduction effect is apparent in this region. The effects in JJA for this region show changes of −25 % OH, +35 % NOx, −12 % O3, and +5 % CO at 900–100 hPa as the most remarkable HR(HO2) effects, as described above. In general, the regional mean effects of HR(HO2) in the North Pacific region are enhanced in JJA, but the mean global effects of HR(HO2) are slightly favored in DJF because of the additional effects of aerosols during this season.
Macintyre and Evans (2011) also found a similar contrast between the behaviors of HR(N2O5) and HR(HO2): the uptake of N2O5 produces both regional and global effects on O3, whereas the uptake of HO2 affects O3 at regional scales more strongly than on a global scale (Macintyre and Evans, 2011). Such features are generally consistent with our results.
3.2.4 Effects of RO2 heterogeneous reactions (HR(RO2))
Effects of HR(RO2) increase the global mean methane lifetime by +0.15 % and change tropospheric abundances of NOx (+0.52 %), O3 (−0.93 %), and CO (−1.78 %) (Table 9). In Fig. 12k–l, significant latitudinal contrasts exist in the NOx changes: large NOx increases at high latitudes and decreases at lower latitudes. These NOx changes probably reflect the reduced formation of PANs, which decreases NOx transport from source regions to remote areas and from the surface to the upper troposphere (Villalta et al., 1996). The model calculated especially large NOx increases (>50 %) for high latitudes around the Arctic Ocean in JJA, indicating a reduction in the formation of PANs (NO PANs), which is linked tightly to the enhanced biogenic emissions of VOCs such as isoprene and terpenes in summer. The PAN-reducing effect of HR(RO2), associated with the suppression of NO oxidation, as seen in the case of HR(HO2), causes a double increase for NOx at the surface as compared to HR(HO2) (144 % in Fig. 12l versus 66 % in Fig. 11l). The increases in NOx in DJF at high latitudes offshore of southern oceans could also reflect reduced transport of NOx under reduced PAN formation caused by HR(RO2) since these coastal areas are located downwind of the major biogenic volatile organic compounds (BVOC) sources of South America, South Africa, and Australia. Moreover, the areas with pronounced NOx increases in Fig. 12k–l are all associated with high-cloud SAD, as seen in Fig. S14 (left panels).
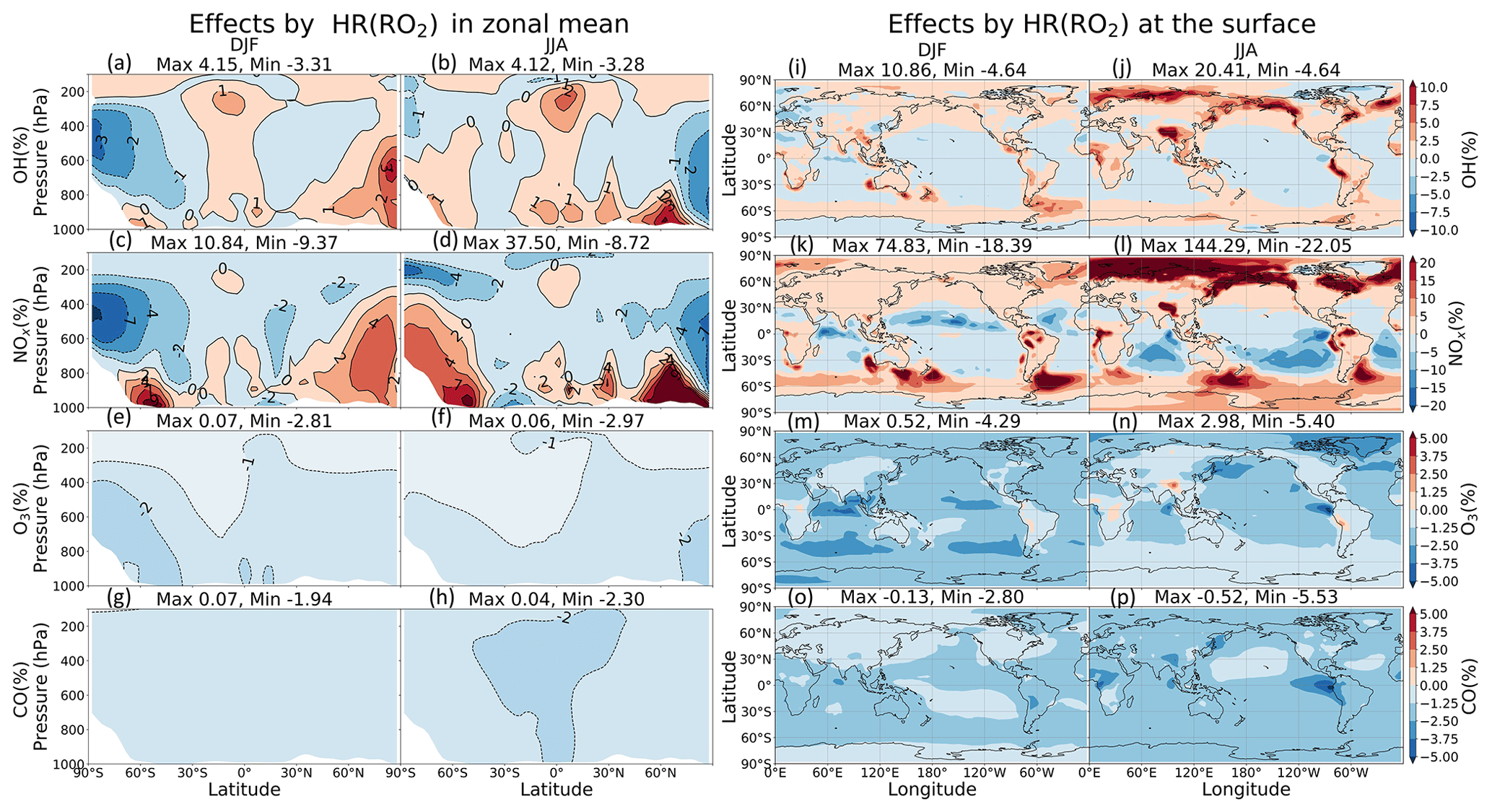
Figure 12Effects of HR(RO2) on both clouds and aerosols in the zonal mean (a–h) and at the surface layer (i–p). Note that the color scale for (a)–(h) is different from that for (i)–(p).

Figure 13Averaged effects of HR(RO2) (a–c) and all HRs (d–f) on OH (first row), NOx (second row), O3 (third row), and CO (fourth row) for each air pressure range. Calculations are for global (a,d), Northern Hemisphere (b, e), and North Pacific region (c, f). Legend is on the first panel for December–January–February (DJF), June–July–August (JJA), and annual (ANN).
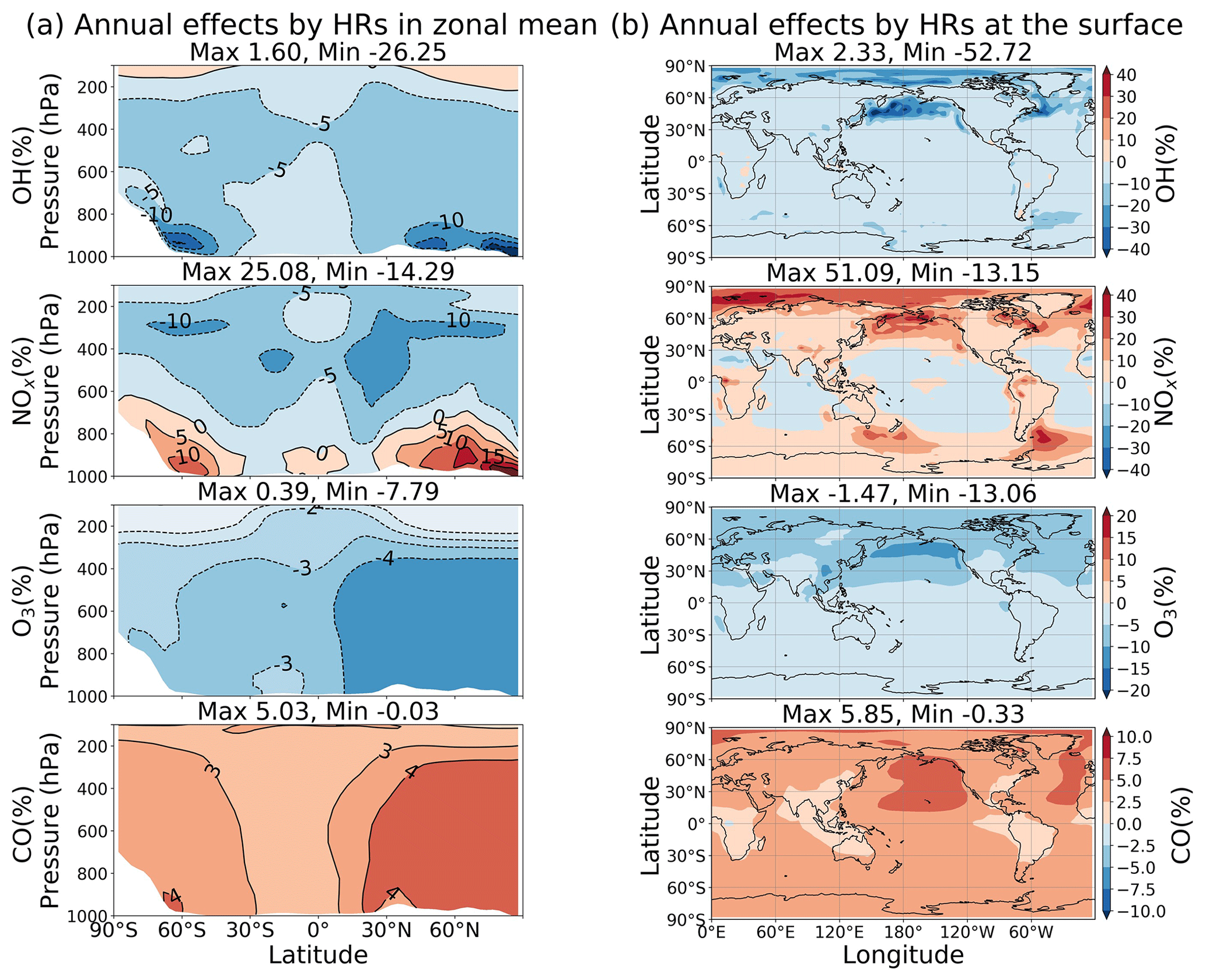
Figure 14Annual zonal mean (a) and surface (b) total HR effects.
In the troposphere, the NO oxidation carried out by RO2 via Reaction (R11) produces another HOx molecule (Reaction R12). Hence, the uptakes of RO2 onto particles are also expected to confine the OH and O3 formations, similar to what the uptake of HO2 does.
However, the different catalytic role of NOx in the oxidizing mechanisms between polluted and remote regions results in different tendencies for OH and O3 (Jacob, 1999). Accordingly, O3 and OH molecules are produced through the oxidation of hydrocarbons in presence of high NOx, whereas HOx molecules are consumed and no O3 is produced in the same process that occurred devoid of NOx. For that reason, less RO2 participating in the hydrocarbon oxidation only reduces OH and O3 levels in polluted regions, e.g., the Chinese region in DJF (Fig. 12i and m), while enhancing OH level and leaving no significant effect on O3 at remote regions, e.g., the NP region (Fig. 12i, j, m, n). The corresponding changes in OH concentration at the surface are in the range of about −5 % to +20 % in JJA (Fig. 12j). O3 levels are slightly reduced throughout the troposphere, at most about −5 % at the surface of the Arctic region above Canada, due to HR(RO2), with only about a 3 % increase, corresponding with the NOx increases at the tropical coasts (Fig. 12n). For CO, different from the increasing impacts by HR(N2O5) and HR(HO2) (+3.42 % and +1.95 %, respectively), a reduction effect occurs through the free troposphere (−1.78 %). CO decrease might be due to reduced secondary CO production from RO2 oxidation ( or ROOH → CO) such as isoprene (Bates and Jacob, 2019) when functionalized RO2 species uptake onto aerosols and clouds particles instead.
The effects of HR(RO2) are primarily attributed to the heterogeneous reaction on clouds rather than on aerosols in terms of changes in NOx and CH4 lifetime, although this cloud effect is far smaller than the cloud effect on the HO2 uptake. The areas with pronounced NOx increases in Fig. 12b are all associated with high-cloud SAD, as seen in Fig. S14 (left panels). Although it is proper to expect the high solubility of RO2 (e.g., CH3O2) from its peroxy substituent (Betterton, 1992; Shepson et al., 1996), it is much less soluble than HO2 because of its lower polarity and thus lower Henry law constant (Jacob, 2000). Consequently, the possible accumulation of CH3O2 in the cloud is attributable to suppression of its gas-phase sink with HO2 (Jacob, 1986).
Figure 13a–c show lat–long means of HR(RO2) effects calculated for the respective pressure ranges: the lat–long values are constrained for the entire globe, the Northern Hemisphere, and North Pacific region. For the entire globe, the contrast effects of HR(RO2) between the lower and higher troposphere on NOx and OH are shown clearly (+3.50 % NOx and +0.55 % OH at 900 hPa, but −2.50 % NOx and −0.75 % OH at 400–500 hPa annually). As a result, the annual mean O3 and CO levels decreased throughout the troposphere, reaching their lowest at −1.60 % O3 and −1.50 % CO at the surface. In JJA, the global effects by HR(RO2) are more concentrated in the lower troposphere, especially in the North Pacific (+3 % OH, +10 % NOx, −3 % O3, −2 % CO at 900–1000 hPa). In DJF, the HR(RO2) effects are observed mostly in the middle and higher troposphere, especially when considering the Northern Hemisphere (−1.25 % OH, −4 % NOx, −2 % O3 at 500–800 hPa).
Table 9Tropospheric abundances changes by HR(N2O5), HR(HO2), HR(RO2), and all HRs for clouds and aerosols.
3.2.5 Total effects of all HRs
As discussed above, different heterogeneous reactions affect tropospheric chemistry differently. However, their effects can either augment or negate others in performing for the atmospheric chemistry. HR(N2O5) is the greatest contributor to reduction of tropospheric OH, O3, and NOx abundances and is more active in the middle troposphere. HR(HO2) reduces OH but increases the abundances of O3 and NOx globally, whereas it exposes a negative effect on O3 level at the surface of the North Pacific region. HR(RO2) similarly has a smaller distribution to the total heterogeneous effects, but its global mean negative effects for O3 are not negligible. The uptake of N2O5 mainly takes place for aerosols, whereas the uptakes of HO2 and RO2 occur more for liquid and ice clouds. Overall, the total effects of all HRs for the whole troposphere are +5.91 % for global mean CH4 lifetime, −2.19 % for NOx (tropospheric abundance), −2.96 % for O3, and +3.28 % for CO (Table 9). At the surface, the annual effects ranged from about −53 % to +2 % for OH, −13 % to +51 % for NOx, −13 % to −2 % for O3, and −0.3 % to +6 % for CO (Fig. 14).
As Fig. 13d–f show for the vertical profiles of HR effects, the change of OH largely concentrated in the lower troposphere (−10 % OH at 900 hPa, calculated for the entire globe) is associated with the HO2 uptake. By contrast, the NOx change is more intensive at higher altitudes (−9 % NOx at 400 hPa, calculated for the entire globe), associated with N2O5 and RO2 uptake. The global mean HR effects on O3 and CO are vertically even, with the highest effects reaching −4 % O3 and +4 % CO at the surface. Globally, HR effects on atmospheric oxidants (OH and O3) are enhanced in DJF because of the higher pollution in the Northern Hemisphere. However, the largest HR effects are apparent for JJA at the surface of the North Pacific (−25 % OH, +38 % NOx, −14 % O3, and +6 % CO as calculated for the 950–1000 hPa layer). These effects are mostly ascribed to HO2 uptake onto clouds. This finding is also apparent from Fig. S15b: these effects reach −66 % for OH, +206 % for NOx, −23 % for O3, and +4.4 % for CO at the surface. They were able to extend up to 400 hPa in the atmosphere. These substantial effects are readily apparent for the large reduction of O3 level during Mirai observation (hatched field in Fig. 4d, T5). However, the major contribution of HR(HO2) to these effects is only partially verified by the ATom1 measurements in this study (Fig.6q, hatched bars at 500–700 hPa). Because of model overestimates of cloud fraction in JJA for the North Pacific region, these effects of HR(HO2) should have existed at a smaller magnitude. For HR effects in the middle to upper troposphere, the N2O5 uptake on aerosols is dominant in these layers and intense in both DJF and JJA.
3.3 Sensitivities of tropospheric chemistry respond to heterogeneous reactions
From the discussion presented above, marked effects of HRs on global tropospheric chemistry are apparent. Here we examine how the tropospheric chemistry responds to the magnitude of HRs' loss rates. To do this, we introduced a factor F for application to the first-order loss rate shown in Eq. (1) for artificially manipulating the HR magnitude.
For this sensitivity test, we only specifically examine HR(HO2) and HR(N2O5) and consider factors of 0–10 for the STD run (Table S1). This test might help to show the effective oxidation sensitivity of the troposphere because future pollution and climate change might enhance the activities of these HRs.
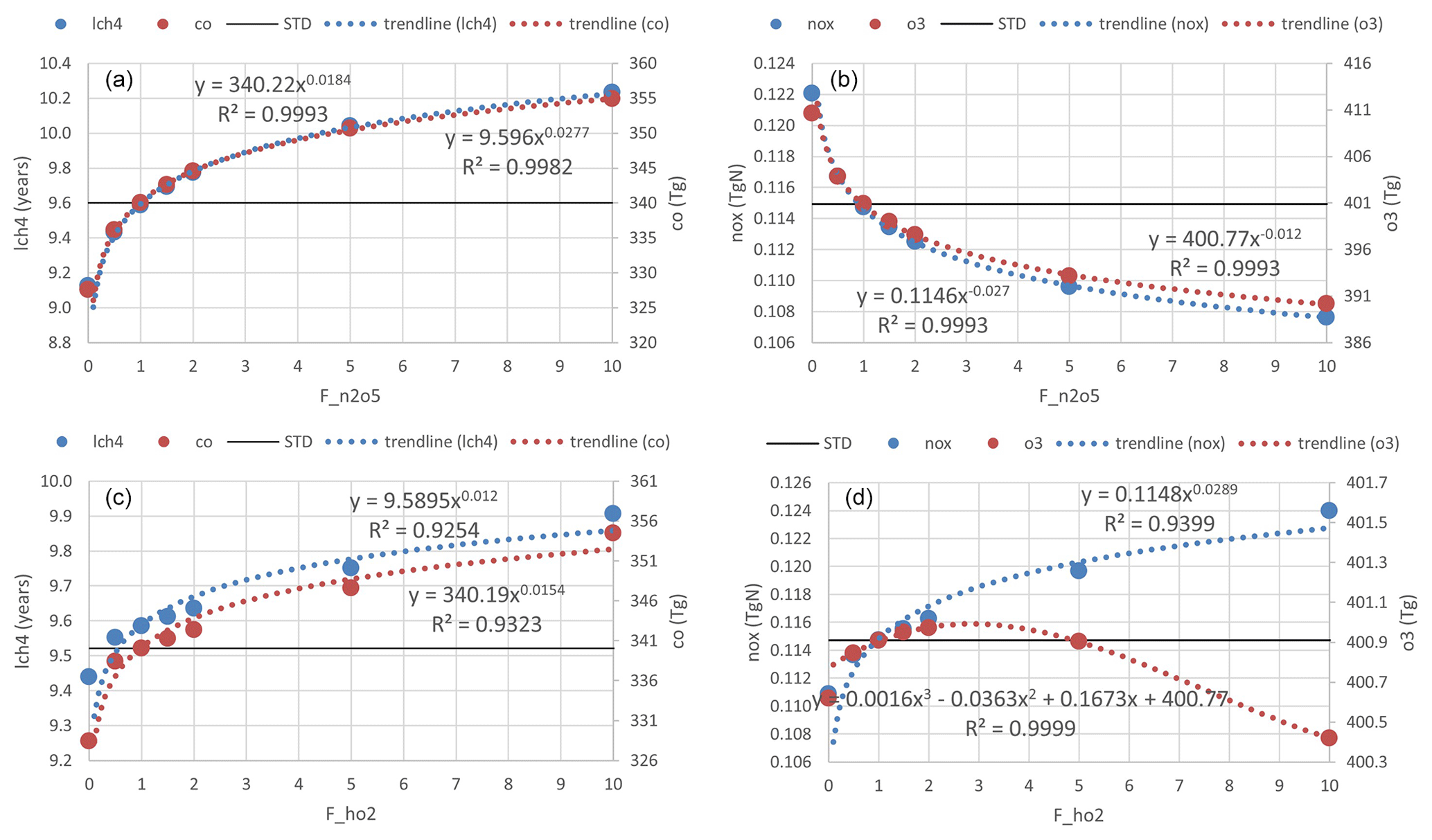
Figure 15Trend lines for the sensitivity of HR(N2O5) (a, b) and HR(HO2) effects (c, d), with uptake rates shown. Panels (a) and (c) show the CH4 lifetime (blue) and tropospheric abundance of CO (red). Panels (b) and (d) show tropospheric abundances of NOx (blue) and O3 (red).
For both effects, we performed nonlinear function fitting with their uptake loss rates, which yielded correlation coefficients higher than 0.93 (Fig. 15). Although both HRs showed negative tendencies for OH and O3 levels, the effect of HR(HO2) on the tropospheric abundance of O3 showed only a small increment with an increasing loss rate (maxima at around F=3) and turned to reduction at higher rates (F>5). As discussed alongside HR(HO2) effects, the O3 level is expected to be primarily reduced only in JJA at the surface of the North Pacific region. At the same time, O3 will be increased gradually elsewhere because of the persistent NOx increment. This behavior produces a positive global mean effect. Figure 10 (dashed lines) shows that manipulation of the HR(HO2) loss rate that is 10 factors higher will effectively increase the negative HR(HO2) effects on O3 in DJF (dashed blue versus solid blue lines, third row, fourth column), which results in a higher tendency of negative values for global mean effects. This sensitivity in DJF might be attributable to the HO2 uptake to aerosols rather than to clouds during this polluted period, which is apparent through comparison of Figs. 11 and S16 for notable events. In DJF, by amplifying the HO2 uptake loss rate by a factor of 10, the effects for the polluted Chinese area (because of HO2 uptake onto aerosols) significantly magnify from −18 % (Fig. 11m) to −47 % (third row, first column in Fig. S16b). In contrast, effects at the surface O3 level in JJA for the North Pacific region (because of HO2 uptake onto clouds) only enhance from −21 % (Fig. 11n) to −29 % (third row, second column in Fig. S16b).
When amplifying HR(N2O5) by a factor of 10, the sensitivities of global effects show no seasonal variation. The HR(N2O5) effects are more sensitive in DJF for the North Pacific region, which link to the higher concentration of aerosol in this season. Otherwise, the HR(N2O5) effects for the generic Northern Hemisphere tend to be more sensitive in JJA as a result of pollutant transportation to the higher troposphere.
Consequently, we suggest that the sensitivity of tropospheric chemistry to HR(N2O5) and HR(HO2) might be attributable to loss activities of aerosols rather than to clouds. The sharply curved effect on O3 because of amplification of HR(HO2) makes sense in plans for ozone pollution control when increased pollution or climate change factors cause the rate of HRs to increase by 3–5 times or more in the future.
The “CHASER” chemistry–climate model was used to investigate global effects of N2O5, HO2, and RO2 uptake. Verification of the model with observations from inland and ocean domains showed adequate agreement for PM2.5, SO, and NO particles and gaseous HNO3, NOx, OH, CO, and O3 concentrations. R, bias, and NRMSE values for SO, NO, and HNO3 at EANET and EMEP stations are comparable with other models. Inclusion of HRs reduced model bias for OH, NO2, CO, and O3, especially in the low troposphere. However, verification with satellite and reanalysis data showed deterioration by HRs for TCO and an overestimate for cloud fraction in the North Pacific region.
The total effects of HRs are important for the tropospheric chemistry that might change +5.91 % CH4 lifetime and −2.19 % NOx, −2.96 % O3, and +3.28 % CO abundances. Global effects are −9 % NOx at 400 hPa, −10 % OH at 900 hPa, and −4 % O3 and +4 % CO at the surface. Global HR effects tend to be enhanced in DJF because of greater amounts of pollution in the Northern Hemisphere.
Total HR effects are contributed mainly by HR(N2O5) onto aerosols in the middle troposphere. At the surface, HR(HO2) is more active and leaves a remarkable disturbance in JJA at the North Pacific region with changes of −70 % for OH, −24 % for O3, +68 % for NOx, and +8 % for CO. These effects were attributed to the uptake of HO2 on cloud particles, which were partially verified with ATom1 observations. HR(RO2), which also favors cloud particles, minorly contributes to the tropospheric chemistry, but it has enormous impacts on PAN and NOx transportation (+144 % NOx for the North Pacific and Atlantic regions in JJA) and the negative changes in CO (−1.78 %), as compared to positive effects by HR(N2O5) and HR(HO2), that can not be neglected. However, the effect magnitude requires further investigation because of model overestimates for cloud fractions in this region.
The sensitivity of tropospheric chemistry to HR magnitude was determined as a nonlinear function. The increasing effect on the global O3 abundance from HR(HO2) will sharply change to a decreasing effect when the uptake rate is amplified by more than 3 times. This turning is ascribed to the uptake onto aerosols in DJF. In general, uptake to aerosols is more responsive to the heterogeneous loss rate than uptake to clouds.
Overall, the N2O5 and HO2 uptakes will sweep away atmospheric oxidants, thereby enhancing concentrations of pollutants. Our results reveal that although HRs are reported to be associated with polluted regions, the global effects of HRs reach further remote regions such as the marine boundary layer at mid-latitudes and the upper troposphere. For ground-based studies of polluted regions such as China, it should be considered that HR(HO2) and HR(RO2) were able to contribute to the NOx increment in DJF and JJA, respectively. Moreover, the HR(HO2) effect might hinder efforts at reducing environmental pollution in urban areas because it increases NOx but decreases O3 at the surface. Therefore, if this reaction is minimized because of a decrease in particulate matter, then the surface ozone level might increase.
The source code for CHASER V4.0 and input data to reproduce results in this work can be obtained from the repository at https://doi.org/10.5281/zenodo.4153452 (Ha et al., 2020).
The underlying data from R/V Mirai cruises for the period 2015–2017 are available from http://www.godac.jamstec.go.jp/darwin/e (last access: 30 June 2020). Due to a recent data security incident, the data owner (JAMSTEC) is suspending public access to this dataset. For any inquiries, please send email to yugo@jamstec.go.jp.
The supplement related to this article is available online at: https://doi.org/10.5194/gmd-14-3813-2021-supplement.
PTMH performed all simulations (except simulations for the cloud fraction validation), interpreted the results, and wrote the manuscript. KS developed the model code, conceived of the presented idea, and supervised the findings of this work and the manuscript preparation. RM carried out the simulations and plots for the validation of cloud fraction. YK and FT provided the R/V Mirai ship data and contributed to the discussion of the work's findings.
The authors declare that they have no conflict of interest.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
We are grateful to the NASA scientists and staff for providing the ATom data (https://espo.nasa.gov/atom/content/ATom, last access: 30 June 2020). The simulations were completed using the supercomputer (NEC SX-Ace and SX-Aurora TSUBASA) at NIES, Japan. The surface observational data for the model validation were taken from the monitoring networks EANET (https://www.eanet.asia/, last access: 25 February 2020) and EMEP (https://www.emep.int/, last access: 25 February 2020).
We also would like to thank the two anonymous reviewers for their helpful suggestions and advice on the earlier draft of the manuscript.
This research was supported by the Global Environment Research Fund (grant nos. S-12 and S-20) of the Ministry of the Environment (MOE), Japan, and by JSPS KAKENHI (grant nos. JP20H04320, JP19H05669, and JP19H04235).
This paper was edited by Gerd A. Folberth and reviewed by two anonymous referees.
Akimoto, H., Nagashima, T., Li, J., Fu, J. S., Ji, D., Tan, J., and Wang, Z.: Comparison of surface ozone simulation among selected regional models in MICS-Asia III – effects of chemistry and vertical transport for the causes of difference, Atmos. Chem. Phys., 19, 603–615, https://doi.org/10.5194/acp-19-603-2019, 2019.
Apodaca, R. L., Huff, D. M., and Simpson, W. R.: The role of ice in N2O5 heterogeneous hydrolysis at high latitudes, Atmos. Chem. Phys., 8, 7451–7463, https://doi.org/10.5194/acp-8-7451-2008, 2008.
Bates, K. H. and Jacob, D. J.: A new model mechanism for atmospheric oxidation of isoprene: global effects on oxidants, nitrogen oxides, organic products, and secondary organic aerosol, Atmos. Chem. Phys., 19, 9613–9640, https://doi.org/10.5194/acp-19-9613-2019, 2019.
Battan, L. J. and Reitan, C. H.: Droplet size measurements in convective clouds, in Artificial simulation of Rain, Pergamon Press, New York, 184–191, 1957.
Betterton, E. A.: Henry's Law constants of soluble and moderately soluble organic gases: effects on aqueous-phase chemistry, in: Gaseous pollutants: Characterization and cycling, Wiley, 24, 1–50, 1992.
Bian, H., Chin, M., Hauglustaine, D. A., Schulz, M., Myhre, G., Bauer, S. E., Lund, M. T., Karydis, V. A., Kucsera, T. L., Pan, X., Pozzer, A., Skeie, R. B., Steenrod, S. D., Sudo, K., Tsigaridis, K., Tsimpidi, A. P., and Tsyro, S. G.: Investigation of global particulate nitrate from the AeroCom phase III experiment, Atmos. Chem. Phys., 17, 12911–12940, https://doi.org/10.5194/acp-17-12911-2017, 2017.
Brown, S. S. and Stutz, J.: Nighttime radical observations and chemistry, Chem. Soc. Rev., 41, 6405–6447, 2012.
Chen, Y., Wolke, R., Ran, L., Birmili, W., Spindler, G., Schröder, W., Su, H., Cheng, Y., Tegen, I., and Wiedensohler, A.: A parameterization of the heterogeneous hydrolysis of N2O5 for mass-based aerosol models: improvement of particulate nitrate prediction, Atmos. Chem. Phys., 18, 673–689, https://doi.org/10.5194/acp-18-673-2018, 2018.
Cooper, P. L. and Abbatt, J. P. D.: Heterogeneous interactions of OH and HO2 radicals with surfaces characteristic of atmospheric particulate matter, J. Phys. Chem., 100, 2249–2254, 1996.
Dentener, F. J.: Heterogeneous chemistry in the troposphere, PhD Thesis, U. of Utrecht, the Netherlands, 1993.
Dentener, F. J. and Crutzen, P. J.: Reaction of N2O5 on tropospheric aerosols: Impact on the global distributions of NOx, O3, and OH, J. Geophys. Res.-Atmos., 98, 7149–7163, 1993.
de Reus, M., Fischer, H., Sander, R., Gros, V., Kormann, R., Salisbury, G., Van Dingenen, R., Williams, J., Zöllner, M., and Lelieveld, J.: Observations and model calculations of trace gas scavenging in a dense Saharan dust plume during MINATROC, Atmos. Chem. Phys., 5, 1787–1803, https://doi.org/10.5194/acp-5-1787-2005, 2005.
Evans, M. J. and Jacob, D. J.: Impact of new laboratory studies of N2O5 hydrolysis on global model budgets of tropospheric nitrogen oxides, ozone, and OH, Geophys. Res. Lett., 32, 1–4, 2005.
Gaudel, A., Cooper, O. R., Ancellet, G., Barret, B., Boynard, A., Burrows, J. P., Clerbaux, C., Coheur, P.-F., Cuesta, J., Cuevas, E., Doniki, S., Dufour, G., Ebojie, F., Foret, G., Garcia, O., Granados-Muñoz, M. J., Hannigan, J. W., Hase, F., Hassler, B., Huang, G., Hurtmans, D., Jaffe, D., Jones, N., Kalabokas, P., Kerridge, B., Kulawik, S., Latter, B., Leblanc, T., Le Flochmoën, E., Lin, W., Liu, J., Liu, X., Mahieu, E., McClure-Begley, A., Neu, J. L., Osman, M., Palm, M., Petetin, H., Petropavlovskikh, I., Querel, R., Rahpoe, N., Rozanov, A., Schultz, M. G., Schwab, J., Siddans, R., Smale, D., Steinbacher, M., Tanimoto, H., Tarasick, D. W., Thouret, V., Thompson, A. M., Trickl, T., Weatherhead, E., Wespes, C., Worden, H. M., Vigouroux, C., Xu, X., Zeng, G., and Ziemke, J.: Tropospheric Ozone Assessment Report: Present-day distribution and trends of tropospheric ozone relevant to climate and global atmospheric chemistry model evaluation, Elem. Sci. Anth., 6, 1–58, 2018.
Geyer, A., Bächmann, K., Hofzumahaus, A., Holland, F., Konrad, S., Klüpfel, T., Pätz, H.-W., Perner, D., Mihelcic, D., Schäfer, H.-J., Volz-Thomas, A., and Platt, U.: Nighttime formation of peroxy and hydroxyl radicals during the BERLIOZ campaign: Observations and modeling studies, J. Geophys. Res.-Atmos., 108, 1–16, 2003.
Ha, T. M. P., Taketani, F., Kanaya, Y., Matsuda, R., and Sudo, K.: Effects of heterogeneous reactions on global tropospheric chemistry (Version CHASER-V4.0) [Code], Zenodo, https://doi.org/10.5281/zenodo.4153452, 2020.
Huijnen, V., Williams, J. E., and Flemming, J.: Modeling global impacts of heterogeneous loss of HO2 on cloud droplets, ice particles and aerosols, Atmos. Chem. Phys. Discuss., 14, 8575–8632, https://doi.org/10.5194/acpd-14-8575-2014, 2014.
Inness, A., Baier, F., Benedetti, A., Bouarar, I., Chabrillat, S., Clark, H., Clerbaux, C., Coheur, P., Engelen, R. J., Errera, Q., Flemming, J., George, M., Granier, C., Hadji-Lazaro, J., Huijnen, V., Hurtmans, D., Jones, L., Kaiser, J. W., Kapsomenakis, J., Lefever, K., Leitão, J., Razinger, M., Richter, A., Schultz, M. G., Simmons, A. J., Suttie, M., Stein, O., Thépaut, J.-N., Thouret, V., Vrekoussis, M., Zerefos, C., and the MACC team: The MACC reanalysis: an 8 yr data set of atmospheric composition, Atmos. Chem. Phys., 13, 4073–4109, https://doi.org/10.5194/acp-13-4073-2013, 2013.
Jacob, D. J.: Chemistry of OH in remote clouds and its role in the production of formic acid and peroxymonosulfate, J. Geophys. Res., 91, 9807–9826, 1986.
Jacob, D. J.: Introduction to Atmospheric Chemistry, Princeton University Press, USA, 1999.
Jacob, D. J.: Heterogeneous chemistry and tropospheric ozone, Atmos. Environ., 34, 2131–2159, 2000.
Janssens-Maenhout, G., Crippa, M., Guizzardi, D., Dentener, F., Muntean, M., Pouliot, G., Keating, T., Zhang, Q., Kurokawa, J., Wankmüller, R., Denier van der Gon, H., Kuenen, J. J. P., Klimont, Z., Frost, G., Darras, S., Koffi, B., and Li, M.: HTAP_v2.2: a mosaic of regional and global emission grid maps for 2008 and 2010 to study hemispheric transport of air pollution, Atmos. Chem. Phys., 15, 11411–11432, https://doi.org/10.5194/acp-15-11411-2015, 2015.
Japan Meteorological Agency/Japan: JRA-55: Japanese 55-year Reanalysis, Daily 3-Hourly and 6-Hourly Data. Research Data Archive at the National Center for Atmospheric Research, Computational and Information Systems Laboratory, https://doi.org/10.5065/D6HH6H41 (last access: 12 June 2020), 2013, updated monthly.
Kanaya, Y., Sadanaga, Y., Hirokawa, J. U. N., Kajii, Y., and Akimoto, H.: Development of a Ground-Based LIF Instrument for Measuring HOx Radicals: Instrumentation and Calibrations, J. Atmos. Chem., 38, 73–110, 2001.
Kanaya, Y., Nakamura, K., Kato, S., Matsumoto, J., Tanimoto, H., and Akimoto, H.: Nighttime variations in HO2 radical mixing ratios at Rishiri Island observed with elevated monoterpene mixing ratios, Atmos. Environ., 36, 4929–4940, 2002a.
Kanaya, Y., Yokouchi, Y., Matsumoto, J., Nakamura, K., Kato, S., Tanimoto, H., Furutani, H., Toyota, K., and Akimoto, H.: Implications of iodine chemistry for daytime HO2 levels at Rishiri Island, Geophys. Res. Lett., 29, 1–4, 2002b.
Kanaya, Y., Kajii, Y., and Akimoto, H.: Solar actinic flux and photolysis frequency determinations byradiometers and a radiative transfer model at Rishiri Island: Comparisons, cloud effects, and detection of an aerosol plumefrom Russian forest fires, Atmos. Environ., 37, 2463–2475, 2003.
Kanaya, Y., Cao, R., Kato, S., Miyakawa, Y., Kajii, Y., Tanimoto, H., Yokouchi, Y., Mochida, M., Kawamura, K., and Akimoto, H.: Chemistry of OH and HO2 radicals observed at Rishiri Island, Japan, in September 2003: Missing daytime sink of HO2 and positive nighttime correlations with monoterpenes, J. Geophys. Res., 112, D11308, https://doi.org/10.1029/2006JD007987, 2007.
Kanaya, Y., Pochanart, P., Liu, Y., Li, J., Tanimoto, H., Kato, S., Suthawaree, J., Inomata, S., Taketani, F., Okuzawa, K., Kawamura, K., Akimoto, H., and Wang, Z. F.: Rates and regimes of photochemical ozone production over Central East China in June 2006: a box model analysis using comprehensive measurements of ozone precursors, Atmos. Chem. Phys., 9, 7711–7723, https://doi.org/10.5194/acp-9-7711-2009, 2009.
Kanaya, Y., Miyazaki, K., Taketani, F., Miyakawa, T., Takashima, H., Komazaki, Y., Pan, X., Kato, S., Sudo, K., Sekiya, T., Inoue, J., Sato, K., and Oshima, K.: Ozone and carbon monoxide observations over open oceans on R/V Mirai from 67∘ S to 75∘ N during 2012 to 2017: testing global chemical reanalysis in terms of Arctic processes, low ozone levels at low latitudes, and pollution transport, Atmos. Chem. Phys., 19, 7233–7254, https://doi.org/10.5194/acp-19-7233-2019, 2019.
Lawrence, M. G. and Crutzen, P. J.: The impact of cloud particle gravitational settling on soluble trace gas distributions, Tellus, Ser. B Chem. Phys. Meteorol., 50B, 263–289, 1998.
Lelieveld, J. and Crutzen, P. J.: Influences of cloud photochemical processes on tropospheric ozone, Nature, 343, 227–233, 1990.
Lelieveld, J. and Crutzen, P. J.: The role of clouds in tropospheric photochemistry, J. Atmos. Chem., 12, 229–267, 1991.
Li, J., Chen, X., Wang, Z., Du, H., Yang, W., Sun, Y., Hu, B., Li, J., Wang, W., Wang, T., Fu, P., and Huang, H.: Radiative and heterogeneous chemical effects of aerosols on ozone and inorganic aerosols over East Asia, Sci. Total Environ., 622–623, 1327–1342, 2018.
Li, K., Jacob, D. J., Liao, H., Shen, L., Zhang, Q., and Bates, K. H.: Anthropogenic drivers of 2013–2017 trends in summer surface ozone in China, P. Natl. Acad. Sci. USA, 116, 422–427, 2019.
Liao, H. and Seinfeld, J. H.: Global impacts of gas-phase chemistry-aerosol interactions on direct radiative forcing by anthropogenic aerosols and ozone, J. Geophys. Res.-Atmos., 110, D18208, https://doi.org/10.1029/2005JD005907, 2005.
Lin, J.-T., Liu, Z., Zhang, Q., Liu, H., Mao, J., and Zhuang, G.: Modeling uncertainties for tropospheric nitrogen dioxide columns affecting satellite-based inverse modeling of nitrogen oxides emissions, Atmos. Chem. Phys., 12, 12255–12275, https://doi.org/10.5194/acp-12-12255-2012, 2012.
Liu, Y. and Wang, T.: Worsening urban ozone pollution in China from 2013 to 2017 – Part 2: The effects of emission changes and implications for multi-pollutant control, Atmos. Chem. Phys., 20, 6323–6337, https://doi.org/10.5194/acp-20-6323-2020, 2020.
Logan, J. A., Prather, M. J., Wofsy, S. C., and McElroy, M. B.: Tropospheric chemistry: a global perspective, J. Geophys. Res., 86, 7210–7254, 1981.
Loukhovitskaya, E., Bedjanian, Y., Morozov, I., and Le Bras, G.: Laboratory study of the interaction of HO2 radicals with the NaCl, NaBr, MgCl2⋅ 6H2O and sea salt surfaces, Phys. Chem. Chem. Phys., 11, 7896–7905, 2009.
Lowe, D., Archer-Nicholls, S., Morgan, W., Allan, J., Utembe, S., Ouyang, B., Aruffo, E., Le Breton, M., Zaveri, R. A., Di Carlo, P., Percival, C., Coe, H., Jones, R., and McFiggans, G.: WRF-Chem model predictions of the regional impacts of N2O5 heterogeneous processes on night-time chemistry over north-western Europe, Atmos. Chem. Phys., 15, 1385–1409, https://doi.org/10.5194/acp-15-1385-2015, 2015.
Macintyre, H. L. and Evans, M. J.: Sensitivity of a global model to the uptake of N2O5 by tropospheric aerosol, Atmos. Chem. Phys., 10, 7409–7414, https://doi.org/10.5194/acp-10-7409-2010, 2010.
Macintyre, H. L. and Evans, M. J.: Parameterisation and impact of aerosol uptake of HO2 on a global tropospheric model, Atmos. Chem. Phys., 11, 10965–10974, https://doi.org/10.5194/acp-11-10965-2011, 2011.
Mao, J., Fan, S., Jacob, D. J., and Travis, K. R.: Radical loss in the atmosphere from Cu-Fe redox coupling in aerosols, Atmos. Chem. Phys., 13, 509–519, https://doi.org/10.5194/acp-13-509-2013, 2013.
Martin, R. V., Jacob, D. J., Yantosca, R. M., Chin, M., and Ginoux, P.: Global and regional decreases in tropospheric oxidants from photochemical effects of aerosols, J. Geophys. Res.-Atmos., 108, 4097, https://doi.org/10.1029/2002JD002622, 2003.
McFarquhar, G. M. and Heymsfield, A. J.: Microphysical characteristics of three anvils sampled during the Central Equatorial Pacific Experiment, J. Atmos. Sci., 53, 2401–2423, 1996.
Monks, P. S., Archibald, A. T., Colette, A., Cooper, O., Coyle, M., Derwent, R., Fowler, D., Granier, C., Law, K. S., Mills, G. E., Stevenson, D. S., Tarasova, O., Thouret, V., von Schneidemesser, E., Sommariva, R., Wild, O., and Williams, M. L.: Tropospheric ozone and its precursors from the urban to the global scale from air quality to short-lived climate forcer, Atmos. Chem. Phys., 15, 8889–8973, https://doi.org/10.5194/acp-15-8889-2015, 2015.
Morgenstern, O., Hegglin, M. I., Rozanov, E., O'Connor, F. M., Abraham, N. L., Akiyoshi, H., Archibald, A. T., Bekki, S., Butchart, N., Chipperfield, M. P., Deushi, M., Dhomse, S. S., Garcia, R. R., Hardiman, S. C., Horowitz, L. W., Jöckel, P., Josse, B., Kinnison, D., Lin, M., Mancini, E., Manyin, M. E., Marchand, M., Marécal, V., Michou, M., Oman, L. D., Pitari, G., Plummer, D. A., Revell, L. E., Saint-Martin, D., Schofield, R., Stenke, A., Stone, K., Sudo, K., Tanaka, T. Y., Tilmes, S., Yamashita, Y., Yoshida, K., and Zeng, G.: Review of the global models used within phase 1 of the Chemistry–Climate Model Initiative (CCMI), Geosci. Model Dev., 10, 639–671, https://doi.org/10.5194/gmd-10-639-2017, 2017.
Morita, A., Kanaya, Y., and Francisco, J. S.: Uptake of the HO2 radical by water: Molecular dynamcs calculations and their implications for atmospheric modeling, J. Geophys. Res.-Atmos., 109, D09201, https://doi.org/10.1029/2003JD004240, 2004.
National Research Council: Rethinking the Ozone Problem in Urban and Regional Air Pollution, The National Academies Press, Washington, D.C., 1991.
Osthoff, H. D., Sommariva, R., Baynard, T., Pettersson, A., Williams, E. J., Lerner, B. M., Roberts, J. M., Stark, H., Goldan, P. D., Kuster, W. C., Bates, T. S., Coffman, D., Ravishankara, A. R., and Brown, S. S.: Observation of daytime N2O5 in the marine boundary layer during New England Air Quality Study – Intercontinental Transport and Chemical Transformation 2004, J. Geophys. Res.-Atmos., 111, D23S14, https://doi.org/10.1029/2006JD007593, 2006.
Platt, U. F., Winer, A. M., Biermann, H. W., Atkinson, R., and Pitts, J. N.: Measurement of Nitrate Radical Concentrations in Continental Air, Environ. Sci. Technol., 18, 365–369, 1984.
Qin, M., Zhongming, C., Hengqing, S., Huan, L., Huihui, W., and Yin, W.: Impacts of heterogeneous reactions to atmospheric peroxides: Observations and budget analysis study, Atmos. Environ., 183, 144–153, 2018.
Qu, Y., Chen, Y., Liu, X., Zhang, J., Guo, Y., and An, J.: Seasonal effects of additional HONO sources and the heterogeneous reactions of N2O5 on nitrate in the North China Plain, Sci. Total Environ., 690, 97–107, 2019.
Richard, W. P.: Chemistry of Atmosphere, Oxford University Press, USA, 2000.
Riemer, N., Vogel, H., Vogel, B., Schell, B., Ackermann, I., Kessler, C., and Hass, H.: Impact of the heterogeneous hydrolysis of N2O5 on chemistry and nitrate aerosol formation in the lower troposphere under photosmog conditions, J. Geophys. Res., 108, 4144, https://doi.org/10.1029/2002JD002436, 2003.
Riemer, N., Vogel, H., Vogel, B., Anttila, T., Kiendler-Scharr, A., and Mentel, T. F.: Relative importance of organic coatings for the heterogeneous hydrolysis of N2O5 during summer in Europe, J. Geophys. Res., 114, D17307, https://doi.org/10.1029/2008JD011369, 2009.
Saathoff, H., Naumann, K.-H., Riemer, N., Kamm, S., Mohler, O., Schurath, U., Vogel, H., and Vogel, B.: The loss of NO2, HNO3, NO3/N2O5, and HO2/HOONO2 on soot aerosol: A chamber and modeling study, Geophys. Res. Lett., 28, 1957–1960, 2001.
Salisbury, G., Rickard, A. R., Monks, P. S., Allan, B. J., Bauguitte, S., Penkett, S. A., Carslaw, N., Lewis, A. C., Creasey, D. J., Heard, D. E., Jacobs, P. J., and Lee, J. D.: Production of peroxy radicals at night via reactions of ozone and the nitrate radical in the marine boundary layer, J. Geophys. Res.-Atmos., 106, 12669–12687, 2001.
Schwartz, S. E.: Mass-Transport Considerations Pertinent to Aqueous Phase Reactions of Gases in Liquid-Water Clouds, in: Chemistry of Multiphase Atmospheric Systems, Springer, Berlin Heidelberg, 415–471, 1986.
Sekiya, T., Miyazaki, K., Ogochi, K., Sudo, K., and Takigawa, M.: Global high-resolution simulations of tropospheric nitrogen dioxide using CHASER V4.0, Geosci. Model Dev., 11, 959–988, https://doi.org/10.5194/gmd-11-959-2018, 2018.
Shepson, P. B., Mackay, E., and Muthuramu, K.: Henry's law constants and removal processes for several atmospheric β-hydroxy alkyl nitrates, Environ. Sci. Technol., 30, 3618–3623, 1996.
Sommariva, R., Bloss, W. J., Brough, N., Carslaw, N., Flynn, M., Haggerstone, A.-L., Heard, D. E., Hopkins, J. R., Lee, J. D., Lewis, A. C., McFiggans, G., Monks, P. S., Penkett, S. A., Pilling, M. J., Plane, J. M. C., Read, K. A., Saiz-Lopez, A., Rickard, A. R., and Williams, P. I.: OH and HO2 chemistry during NAMBLEX: roles of oxygenates, halogen oxides and heterogeneous uptake, Atmos. Chem. Phys., 6, 1135–1153, https://doi.org/10.5194/acp-6-1135-2006, 2006.
Stadtler, S., Simpson, D., Schröder, S., Taraborrelli, D., Bott, A., and Schultz, M.: Ozone impacts of gas–aerosol uptake in global chemistry transport models, Atmos. Chem. Phys., 18, 3147–3171, https://doi.org/10.5194/acp-18-3147-2018, 2018.
Sudo, K. and Akimoto, H.: Global source attribution of tropospheric ozone: Long-range transport from various source regions, J. Geophys. Res.-Atmos., 112, D12302, https://doi.org/10.1029/2006JD007992, 2007.
Sudo, K., Takahashi, M., Kurokawa, J. I., and Akimoto, H.: CHASER: A global chemical model of the troposphere 1. Model description, J. Geophys. Res.-Atmos., 107, ACH 7-1–ACH 7-20, 2002.
Takemura, T., Okamoto, H., Maruyama, Y., Numaguti, A., Higurashi, A., and Nakajima, T.: Global three-dimensional simulation of aerosol optical thickness distribution of various origins, J. Geophys. Res., 105, 17853–17873, 2000.
Taketani, F., Kanaya, Y., and Akimoto, H.: Kinetics of heterogeneous reactions of HO2 radical at ambient concentration levels with (NH4)2SO4 and NaCl aerosol particles, J. Phys. Chem. A, 112, 2370–2377, 2008.
Taketani, F., Kanaya, Y., and Akimoto, H.: Heterogeneous loss of HO2 by KCl, synthetic sea salt, and natural seawater aerosol particles, Atmos. Environ., 43, 1660–1665, 2009.
Taketani, F., Kanaya, Y., Pochanart, P., Liu, Y., Li, J., Okuzawa, K., Kawamura, K., Wang, Z., and Akimoto, H.: Measurement of overall uptake coefficients for HO2 radicals by aerosol particles sampled from ambient air at Mts. Tai and Mang (China), Atmos. Chem. Phys., 12, 11907–11916, https://doi.org/10.5194/acp-12-11907-2012, 2012.
Thornton, J. A., Jaeglé, L., and McNeill, V. F.: Assessing known pathways for HO2 loss in aqueous atmospheric aerosols: Regional and global impacts on tropospheric oxidants. J. Geophys. Res.-Atmos., 113, D05303, https://doi.org/10.1029/2007JD009236, 2008.
Tie, X., Brasseur, G., Emmons, L., Horowitz, L., and Kinnison, D.: Effects of aerosols on tropospheric oxidants: A global model study, J. Geophys. Res.-Atmos., 106, 22931–22964, 2001.
Villalta, P. W., Lovejoy, E. R., and Hanson, D. R.: Reaction probability of peroxyacetyl radical on aqueous surfaces, Geophys. Res. Lett., 23, 1765–1768, 1996.
Wang, H., Lu, K., Guo, S., Wu, Z., Shang, D., Tan, Z., Wang, Y., Le Breton, M., Lou, S., Tang, M., Wu, Y., Zhu, W., Zheng, J., Zeng, L., Hallquist, M., Hu, M., and Zhang, Y.: Efficient N2O5 uptake and NO3 oxidation in the outflow of urban Beijing, Atmos. Chem. Phys., 18, 9705–9721, https://doi.org/10.5194/acp-18-9705-2018, 2018.
Wang, Z., Wang, W., Tham, Y. J., Li, Q., Wang, H., Wen, L., Wang, X., and Wang, T.: Fast heterogeneous N2O5 uptake and ClNO2 production in power plant and industrial plumes observed in the nocturnal residual layer over the North China Plain, Atmos. Chem. Phys., 17, 12361–12378, https://doi.org/10.5194/acp-17-12361-2017, 2017.
Watanabe, S., Hajima, T., Sudo, K., Nagashima, T., Takemura, T., Okajima, H., Nozawa, T., Kawase, H., Abe, M., Yokohata, T., Ise, T., Sato, H., Kato, E., Takata, K., Emori, S., and Kawamiya, M.: MIROC-ESM 2010: model description and basic results of CMIP5-20c3m experiments, Geosci. Model Dev., 4, 845–872, https://doi.org/10.5194/gmd-4-845-2011, 2011.
Xia, M., Wang, W., Wang, Z., Gao, J., Li, H., Liang, Y., Yu, C., Zhang, Y., Wang, P., Zhang, Y., Bi, F., Cheng, X., and Wang, T.: Heterogeneous Uptake of N2O5 in Sand Dust and Urban Aerosols Observed during the Dry Season in Beijing, Atmosphere, 10, 204, https://doi.org/10.3390/atmos10040204, 2019.
Zheng, B., Tong, D., Li, M., Liu, F., Hong, C., Geng, G., Li, H., Li, X., Peng, L., Qi, J., Yan, L., Zhang, Y., Zhao, H., Zheng, Y., He, K., and Zhang, Q.: Trends in China's anthropogenic emissions since 2010 as the consequence of clean air actions, Atmos. Chem. Phys., 18, 14095–14111, https://doi.org/10.5194/acp-18-14095-2018, 2018.