the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 13 Jun 2024
| 13 Jun 2024
Incorporating Oxygen Isotopes of Oxidized Reactive Nitrogen in the Regional Atmospheric Chemistry Mechanism, version 2 (ICOIN-RACM2)
Wendell W. Walters
Masayuki Takeuchi
Nga L. Ng
Meredith G. Hastings
The oxygen isotope anomaly (Δ17O = δ17O − 0.52 × δ18O > 0) has proven to be a robust tool for probing photochemical cycling and atmospheric formation pathways of oxidized reactive nitrogen (NOy). Several studies have developed modeling techniques to implicitly model Δ17O of NOy molecules based on numerous assumptions that may not always be valid. Thus, these models may be oversimplified and limit our ability to compare model Δ17O values of NOy with observations. In this work, we introduce a novel method for explicitly tracking Δ17O transfer and propagation into NOy and odd oxygen (Ox), integrated into the Regional Atmospheric Chemistry Mechanism, version 2 (RACM2). Termed ICOIN-RACM2 (InCorporating Oxygen Isotopes of NOy in RACM2), this new model includes the addition of 55 new species and 729 replicate reactions to represent the propagation of Δ17O derived from O3 into NOy and Ox. Employing this mechanism within a box model, we simulate Δ17O for various NOy and Ox molecules for chamber experiments with varying initial nitrogen oxides (NOx = NO + NO2) and α-pinene conditions, revealing response shifts in Δ17O linked to distinct oxidant conditions. Furthermore, diel cycles are simulated under two summertime scenarios, representative of an urban and rural site, revealing pronounced Δ17O diurnal patterns for several NOy components and substantial Δ17O differences associated with pollution levels (urban vs. rural). Overall, the proposed mechanism offers the potential to assess NOy oxidation chemistry in chamber studies and air quality campaigns through Δ17O model comparisons against observations. The integration of this mechanism into a 3-D atmospheric chemistry transport model is expected to notably enhance our capacity to model and anticipate Δ17O across landscapes, consequently refining model representations of atmospheric chemistry and tropospheric oxidation capacity.
- Article
(2437 KB) - Full-text XML
-
Supplement
(833 KB) - BibTeX
- EndNote





Nitrogen oxides (NOx = NO + NO2) are essential trace gases primarily released through human activities, carrying significant implications for air quality, nutrient deposition, and the climate system (Galloway et al., 2004; Pinder et al., 2012). NOx directly modulates atmospheric oxidation processes, consequently impacting the concentrations of various trace gases, including greenhouse gases (Prinn, 2003). Ultimately, NOx is removed from the atmosphere as atmospheric nitrate. This global process is dominated by the formation of inorganic nitrate, encompassing nitric acid (HNO3) and particulate nitrate (pNO3) (Alexander et al., 2020), although the generation of organic nitrates (RONO2) might be significant in remote and rural areas (Browne and Cohen, 2012). However, both pNO3 and RONO2 may not be a terminal sink for NOx due to the potential for renoxification from photolysis (Wang et al., 2023; Gen et al., 2022). Uncertainties surrounding the rate of NOx oxidation to atmospheric nitrate constitute a substantial source of ambiguity in models, influencing ozone (O3) and hydroxyl radical (OH) formation, with important implications for greenhouse gas removal rates (Newsome and Evans, 2017).
The oxygen mass-independent fractionation leading to an oxygen isotope anomaly (Δ17O = δ17O − 0.52 × δ18O > 0) has emerged as a potent tool for evaluating the photochemical cycling and oxidation chemistry of NOx and its oxidized products (NOy = NOx + HNO3 + RONO2 + nitrous acid (HONO) + peroxyacetyl nitrate (PAN) + etc.) (Alexander et al., 2020, 2009; Hastings et al., 2003; Michalski et al., 2003; Morin et al., 2011; Walters et al., 2019). While several atmospheric reactions can induce oxygen mass-independent fractionation (Röckmann et al., 1998; Velivetskaya et al., 2016), O3 is the overwhelming source of mass-independent fractionation in the lower atmosphere, which derives from unconventional isotope effects during its formation (Gao and Marcus, 2001). In this work, we focus on the propagation of the oxygen isotope anomaly from O3 mass-independent fractionation into NOy and Ox molecules for applications to the lower atmosphere. The Δ17O(O3) has been measured to be between 20 ‰ and 46 ‰ (Krankowsky et al., 2000; Mauersberger et al., 2001). This range of values has been shown to track with the pressure and temperature associated with O3 formation (Thiemens and Jackson, 1990; Morton et al., 1990). For typical tropospheric conditions, O3 exhibits a Δ17O between 20 ‰ and 30 ‰ (Johnston and Thiemens, 1997), with recent near-surface observations suggesting a mean Δ17O(O3) near 26 ‰ (Vicars and Savarino, 2014; Vicars et al., 2012; Ishino et al., 2017). O3 is also isotopically asymmetrical such that the Δ17O of its terminal and central O atoms are different (Janssen, 2005; Marcus, 2008). This intramolecular Δ17O distribution is significant because the terminal O atom of O3 (defined as ) is preferentially transferred during oxidation reactions involving O3 (Bhattacharya et al., 2008; Liu et al., 2001; Michalski and Bhattacharya, 2009; Walters and Michalski, 2016). The relationship between Δ17O(O3) and Δ17O() is complex, though experimental data has suggested the following relationship:
Applying this relationship to the assumed tropospheric mean Δ17O(O3) of 26 ‰ would imply a of 39 ‰, which is near the average of recent near-surface observations of 39.3 ± 2 ‰ (Vicars and Savarino, 2014). It is important to note that there could be seasonal differences in as inferred from Δ17O measurements of nitrate at Dome C (Savarino et al., 2016). On the other hand, direct observations of have reported insignificant seasonal variability at Dumont d'Urville (Ishino et al., 2017). Stratospheric intrusion events could introduce O3 with an elevated due to higher stratosphere values relative to the troposphere (Krankowsky et al., 2007). Nevertheless, a recent modeling study of Δ17O of atmospheric nitrate indicated that an assumed value of 39 ‰, reasonably reproduced global tropospheric observations (Alexander et al., 2020). Further, recent chamber simulations have reported a Δ17O(NO2) that reached as high as 41.2 ‰ (Blum et al., 2023), which is within the measurement uncertainty of the assumed value of 39.3 ± 2 ‰, assuming NO2 formation to be dominated by NO reaction with O3. Thus, while there may be some unresolved uncertainty regarding the value, an assumed tropospheric average of 39.3 ± 2 ‰ should reasonably approximate Δ17O propagation into NOy molecules in the lower troposphere. In contrast, most other oxygen-bearing atmospheric molecules, such as oxygen (O2), water (H2O), and peroxy radicals (RO2 or HO2), possess (or are expected to possess) Δ17O values near 0 ‰ (Lyons, 2001). These large Δ17O differences enable the quantitative tracking of the influence of O3 in NOx oxidation chemistry.
Past observations of Δ17O in atmospheric nitrate, which includes HNO3, pNO3, and wet-deposited nitrate (), have generally shown marked seasonal variations, reflecting shifts between O3 and HOx chemical regimes influencing NOx photochemical cycling and atmospheric nitrate production (Kim et al., 2023; Michalski et al., 2012, 2003). However, harnessing the full diagnostic potential of Δ17O observations necessitates a model framework that can accurately assess and refine the representation of nitrate chemistry while linking it to nitrogen deposition and air quality. Several 0-D box models and a single 3-D global atmospheric chemistry model have been developed to simulate Δ17O (Michalski et al., 2003; Morin et al., 2011; Alexander et al., 2020, 2009). These models often rely on implicit tagging of NO2 and HNO3 production rates, underpinned by assumptions regarding oxygen isotope mass balance calculations, NOx photochemical cycling dynamics, and Δ17O values of reactive oxygen species (Ox).
Table 1Summary of the major formation pathways of several NOy components, reaction types, and their expected Δ17O values based on oxygen isotope mass balance. X refers to halogens (Br, Cl, and I), and HC refers to hydrocarbons.

∗ Δ17O calculated from the nitrooxy (-NO3) functional group.
While most existing Δ17O measurements pertain to atmospheric nitrate from deposition and filter samples, our capability to measure Δ17O in other NOy molecules has been rapidly expanding (Albertin et al., 2021; Blum et al., 2023). Based on oxygen isotope mass balance principles, substantial Δ17O variations are anticipated among different NOy molecules, including NO2, HONO, peroxy nitrates (RO2NO2), organic nitrates (RONO2), and HNO3, contingent upon their formation pathways (Table 1). These mass balance considerations necessitate precise knowledge of the Δ17O values for several NOy and Ox molecules. Conventional model approaches have assumed that Δ17O of OH, RO2, and HO2 are approximately equal to 0 ‰ due to water vapor isotope exchange or transfer of O atoms from atmospheric O2 (Michalski et al., 2012; Barkan and Luz, 2003). However, some of these assumptions are not valid for all relevant atmospheric conditions, such as under low relative humidity and high NOx conditions, in which the chemical reactivity of OH could be higher than its chemical lifetime to achieve isotope equilibrium with H2O (Michalski et al., 2012). Further, Δ17O(NO) is commonly assumed to be equal to Δ17O(NO2) due to its rapid photochemical cycling, such that the Δ17O values of NO and NO2 reflect the relative contributions of the oxidants involved in NOx photochemical cycling (Alexander et al., 2020, 2009; Michalski et al., 2003; Morin et al., 2011). However, recent diel observations of δ18O(NO2) (which tracks with Δ17O) and Δ17O(NO2) reveal that this assumption is not universally valid due to substantial nocturnal NO emissions close to the surface (Walters et al., 2018; Albertin et al., 2021). The freshly emitted NO, with a presumed Δ17O of 0 ‰, would dilute the residual Δ17O of NOx from the daytime. The nocturnal primary emissions of NOy components, including NO, NO2, and HONO significantly impacts our ability to model Δ17O using implicit methods in polluted regions, employing prior modeling techniques and oxygen isotope mass balance calculations. This modeling limitation presently impedes our capacity to leverage models for comparison with Δ17O observational constraints quantitatively to improve understanding of regional and global NOx oxidation chemistry.
This study is dedicated to addressing uncertainties in modeling Δ17O for various NOy molecules. We introduce a novel gas-phase chemical mechanism, designated “InCorporating Oxygen Isotopes of NOy in RACM2”, built upon the foundation of the Regional Atmospheric Chemistry Model, version 2 (RACM2) (Goliff et al., 2013). This innovative mechanism explicitly traces the transfer and propagation of Δ17O from O3 into NOy molecules, with important future implications for chamber experiments and air quality studies.
2.1 ICOIN-RACM2 description
The Incorporating Oxygen Isotopes of Oxidized Reactive Nitrogen in the Regional Atmospheric Chemistry Mechanism, version 2 (ICOIN-RACM2), was based on the widely used RACM2 gas-phase chemical mechanism framework (Goliff et al., 2013). The RACM2 mechanism was developed to be able to simulate remote to polluted conditions from the surface to the upper troposphere. The mechanism includes 46 reactions to represent inorganic chemistry. The mechanism aggregates organic reactions based on the magnitude of emission rates, similarities in functional groups, and the compounds' reactivity (Stockwell et al., 1997) and consists of 54 stable organic species, 42 organic intermediates, 317 reactions, including 24 photolysis reactions. Overall, the RACM2 simulated concentrations of gas-phase products compare favorably to environmental chamber data (Goliff et al., 2013).
Table 2Summary of the considered O exchange reactions and reaction rates in the ICOIN-RACM2 mechanism. These reactions were adapted from Lyons (2001).

To simulate Δ17O in various NOy and Ox molecules, the transfer and propagation of the oxygen isotope anomaly deriving from O3 were explicitly modeled in the employed chemical mechanism. Previously, a study developed a kinetic model that explicitly tracks the 16O, 17O, and 18O abundance involving NOx/O3/O2 reactions (Michalski et al., 2014). Here, we have adapted and simplified this model framework to explicitly track the transfer and propagation of O atoms derived from the terminal end of O3 without simulating and tracking the absolute 16O, 17O, and 18O abundances, which can be tedious to employ in a detailed chemical mechanism. Our approach tagged O atoms transferred from O3 as “Q” and tracked the interactions and propagation of “Q” among NOy and Ox isotopologues using mass balance and considering isotopologue reaction stoichiometry. The tagging of O isotopologues was not conducted for large O reservoirs, including O2 and H2O. The reaction mechanism involved reactions with a single tagged O isotopologue, in which one tagged O isotopologue compound was found in the reactant and product, for example, NO + O3 → NOQ + O2 and NQ + O3 → NQ2 + O2. Additionally, the mechanism involved reactions containing multiple tagged O compounds in the reactants and products, for example, NQ + NO3 → NOQ + NO2. For these, multiple O-tagged isotopologue reactions, statistical probabilities, and mass balance were considered in the product distributions. The explicit tracking and propagation of “Q” in the ICOIN-RACM2 lead to the renaming of 19 reactions, the addition of 729 reactions replicated for the considered O isotopologues, and the addition of 55 oxygen isotopologues of NOy and Ox relative to RACM2. Additionally, 26 oxygen isotope exchange reactions were added to the ICOIN-RACM2 chemical mechanism (Lyons, 2001) (Table 2).
Based on the model output of the concentrations of the oxygen isotopologues, the Δ17O of various NOy and HOx molecules were calculated as follows (Eq. 2):
where X refers to the various NOy and Ox molecules and f(Q) is the fractional amount of O atoms deriving from O3 for a particular molecule (i.e., the fractional amount of “Q” atoms). The Δ17O() represents the Δ17O value of the terminal and transferable O atom of O3. For the demonstration of the developed mechanism for applications to chamber simulations and tropospheric chemistry, we have utilized a constant value of 39.3 ± 2 ‰ based on near-surface-level collections of O3 on a nitrite coated filter (Vicars and Savarino, 2014; Ishino et al., 2017). This value was recently utilized in the global modeling of Δ17O of atmospheric nitrate, demonstrating reasonable agreement between model simulation and observations of tropospheric nitrate (Alexander et al., 2020). The could have temporal variability and be influenced by stratospheric intrusion events, which could introduce O3 with a higher value. The developed model framework is highly flexible, and the user may apply a different than chosen for our model simulations, which will allow users to investigate both the chemical and variability on Δ17O of NOy and Ox species when interpreting field observations. The f(Q) for the various considered molecules is calculated as follows (Eq. 3):
where Z represents the oxygen isotopologues of molecule X, which can contain or not contain Q (Zwith iQ); i represents the number of Q isotopes present in each Z of X; and j represents the maximum number of Q isotopes that can exist in X. Overall, this equation considers the distribution of Q isotopes within different arrangements and calculates the fraction of Q isotopes in the molecule relative to the total number of oxygen atoms. While our mechanism and application is focused on evaluating the propagation of oxygen isotope mass-independent fractionation from O3 into NOy and Ox, the model could be adapted for tracking other potential oxygen mass-independent fractionation, such as HO2 + HO2 or CO + OH reactions (Röckmann et al., 1998; Velivetskaya et al., 2016), by adjusting the product distribution of “Q” and “O”, such that the fraction of “Q” once scaled by the chosen value would match the intended Δ17O value associated with the oxygen mass-independent fractionation. Previous experiments have reported an increase in Δ17O(H2O2) as the initial O2 concentrations increased (Velivetskaya et al., 2016). This result was concluded to reflect the increased role of O3 reactions in H2O2 formation, which is already tracked in our mechanism. The CO + OH reaction, producing a Δ17O in the residual CO, would be extremely unlikely to affect the Δ17O of NOy or Ox due to the long atmospheric lifetime of CO relative to NOy or Ox. Therefore, we did not explicitly test these reactions' influence on Δ17O of NOy or Ox in this work but could easily be adapted in future iterations of the model.
The RACM2 mechanism is a gas-phase mechanism and does not include heterogeneous reactions, which could limit the ICOIN-RACM2 mechanism's ability to accurately simulate Δ17O values, particularly of HONO and HNO3 (Table 1). Gas-phase mechanisms are often used in larger chemical transport models that also include aerosol modules to calculate heterogeneous chemistry reaction rates. When utilizing ICOIN-RACM2 to simulate Δ17O values (and RACM2 for simulating concentrations) in box models that lack aerosol modules, appropriate reactions should be included using pseudo-first-order reaction rate constants to calculate heterogeneous hydrolysis. However, estimating the heterogeneous reaction rates is not trivial and depends on the molecular speed; uptake coefficients, which depend on aerosol chemical composition; and surface area density. These reaction rates may need to be treated in a case-by-case circumstance. Since the ICOIN-RACM2 mechanism does not model particulate nitrate, we cannot model its photolysis, which could limit our ability to simulate Δ17O(HONO). Additionally, our gas-phase mechanism does not include NO2 heterogeneous reactions, which could also be an important source of HONO (Chai et al., 2021). Users interested in accurately simulating Δ17O(HONO) may need to consider adding relevant reactions. Still, a future comparison between Δ17O(HONO) observations and model simulations based on the ICOIN-RACM2 framework should provide pivotal insight into HONO formation.
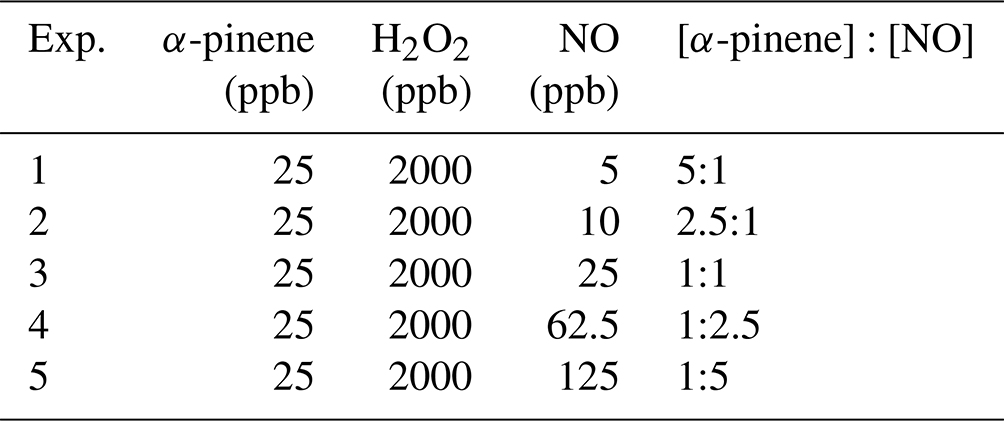
Table 3Summary of the precursor concentrations for the box model simulations of α-pinene and NO photochemical oxidation chamber experiments. All experiments were simulated at a fixed temperature and relative humidity of 22 °C and 1 %, respectively.
2.2 Box model description
The ICOIN-RACM2 mechanism was utilized in the Framework for 0-D Atmospheric Modeling (F0AM) box model (Wolfe et al., 2016). This box model presents a high degree of flexibility, allowing it to be seamlessly adapted for a wide range of simulation scenarios, and can perform online computation of photolysis frequencies. The ICOIN-RACM2 mechanism was developed for use in the F0AM. In this work, the F0AM model was utilized to illustrate the capacity of the ICOIN-RACM2 mechanism for simulating Δ17O values from photochemical chamber experiments and steady-state diel cycles.
2.2.1 Chamber simulations
Box model simulations were conducted to evaluate α-pinene and NOx chemistry under various initial conditions that included variable [α-pinene]:[NOx] ratios (Table 3). These simulations were conducted using similar initial volatile organic compound (VOC) and H2O2 levels utilized in recently conducted chamber experiments (Takeuchi and Ng, 2019). We have also varied the initial VOC and NOx concentration levels to look at the impact of changing initial conditions and model chemistry on Δ17O values. As α-pinene is an important monoterpene, and its oxidation in the presence of NOx constitutes an important mechanism of coupled biogenic–anthropogenic interaction, it has important consequences for air quality, climate, global reactive nitrogen budget, and secondary organic aerosols (SOAs) (Romer et al., 2016; Zare et al., 2018; Ng et al., 2017). The model was initiated for each experiment using NO, α-pinene, and H2O2 (OH precursor) concentrations. The α-pinene and H2O2 concentrations were fixed at 25 and 2000 ppb, respectively, while the initial NO concentrations were varied from 5 to 125 ppb to simulate oxidation chemistry in a range of [α-pinene]:[NOx] conditions (Table 3). The pressure, temperature, and relative humidity were fixed at 1013 mbar, 295 K, and 1 %, respectively. The measured chamber light flux data from the Georgia Institute of Technology, Environmental Chamber Facility was also utilized.
The model was run for 4 h for each simulated experiment. Both gas and particle chamber wall loss were not considered in the chamber simulation comparison. Monoterpene organic nitrate hydrolysis can be an important loss process and formation pathway of HNO3 (Zare et al., 2018; Fisher et al., 2016; Takeuchi and Ng, 2019; Wang et al., 2021) but was not considered in the model because of the low relative humidity conditions. Additionally, heterogeneous pathways leading to the production of HNO3, such as N2O5, were not included. For the hypothetical simulations, this should not impact the reliability of the predictions due to the photochemical conditions of the simulated chamber experiments, low relative humidity, and high organic carbon content of produced particles, which would be reasonably expected to lead to a low N2O5 uptake coefficient (Escorcia et al., 2010). When utilizing the ICOIN-RACM2 mechanism to simulate chamber experimental Δ17O data, gas and particle wall loss, organic nitrate hydrolysis, and NOy heterogeneous reactions should be considered, but it will depend on the chamber and reaction conditions and should be treated in a case-by-case circumstance. The model simulations evaluated the Δ17O temporal variation of NO2, HONO, monoterpene-derived organic nitrate (ONIT), HNO3, OH, and HO2 and investigated their changes in response to the experimental oxidant conditions.
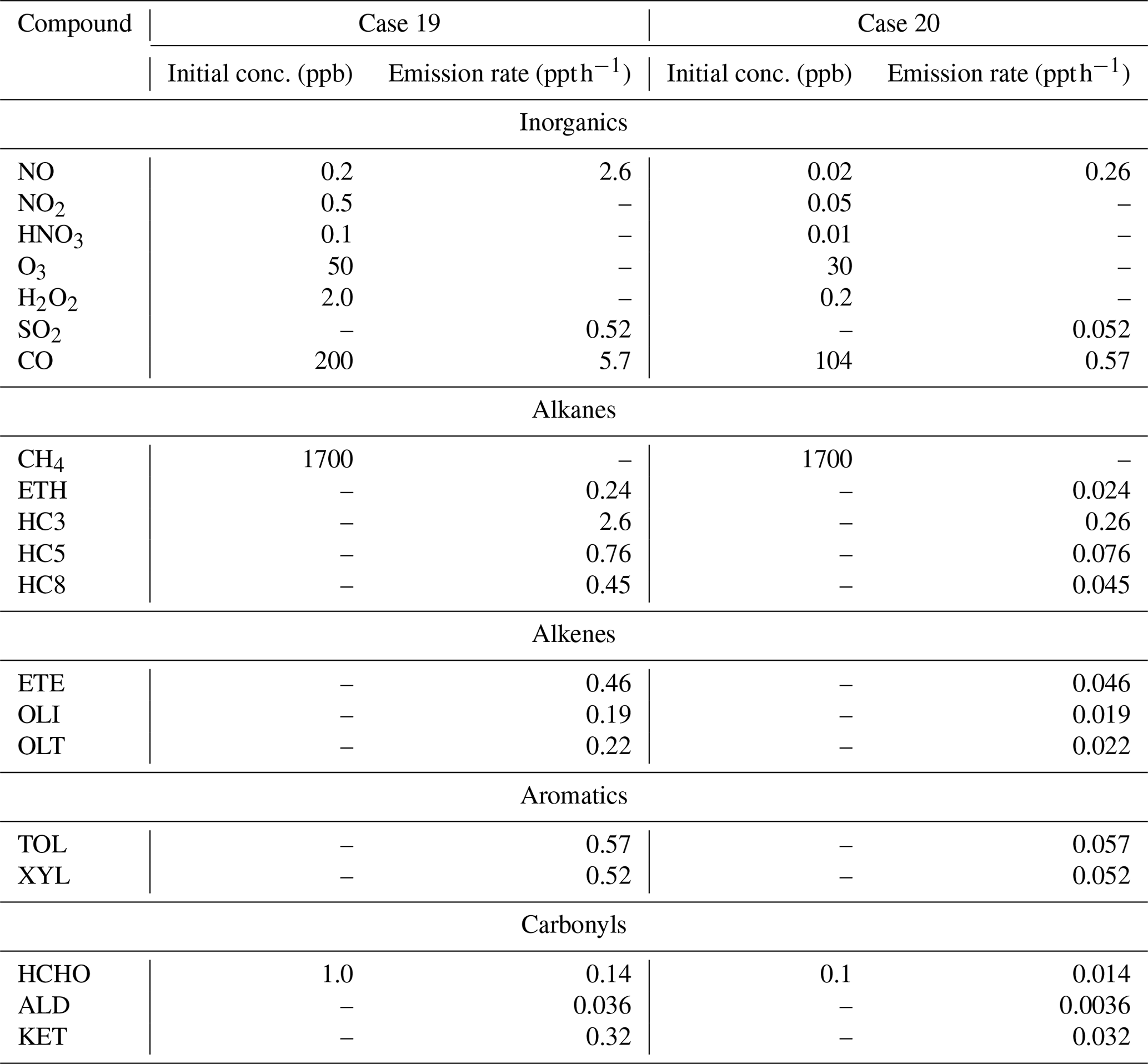
Table 4Conditions for the box model simulations of the diel cycle for two summertime scenarios. The simulations were conducted at a fixed elevation of 0 km, temperature of 298 K, pressure of 1013.25 mbar, and on 21 June 2015, in Providence, RI (41.82° N, 71.41° W). The scenarios were adapted from Stockwell et al. (1997).
2.2.2 Diel variations
Box model simulations were also conducted in steady-state diel cycles for two summertime scenarios. These scenarios (Case 19 and Case 20) were based on previous case studies utilized to evaluate the RACM and RACM2 mechanism (Stockwell et al., 1997; Goliff et al., 2013). Briefly, Case 19 represents a somewhat polluted atmosphere with emissions of NOx and organic compounds, and Case 20 represents a relatively clean atmosphere with the initial concentrations and emission rates of NOx and organic compounds reduced by a factor of 10 (Table 4). These scenarios would be analogous to near-surface summertime environments at an urban (i.e., Case 19) and rural (i.e., Case 20) environment. The box model simulations were conducted for the initial conditions and with a fixed elevation of 0 km, temperature of 298 K, pressure of 1013.25 mbar, and for 21 June as previously described (Stockwell et al., 1997). The simulations were conducted for Providence, RI (41.82° N, 71.41° W), and the diel photolysis rates were calculated using the online module in F0AM (Wolfe et al., 2016). To avoid the buildup of concentrations in the box model, a dilution lifetime of 24 h was incorporated into the simulations, as previously described (Wolfe et al., 2016). The model simulations were run for 5 d at a 1 h interval. The first 2 d of the simulation were used as a spin-up period, and the diel cycles were evaluated based on the average of the final 3 d of the simulation.
Table 5Summary of the NOy heterogeneous reactions, assumed uptake coefficients, and the reference or calculated pseudo-first-order reaction rates adapted in the RACM2(het) and ICOIN-RACM2(het) chemical mechanisms.
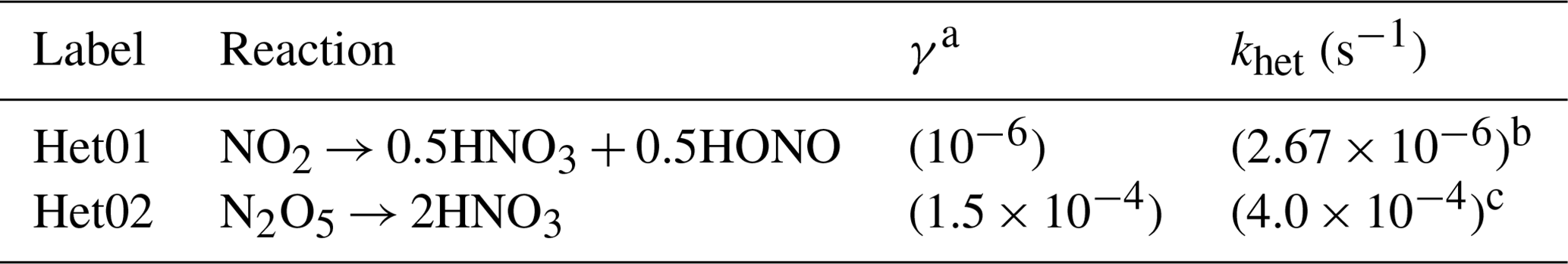
a Adapted from Holmes et al. (2019). b Calculated by scaling the khet (N2O5) based on the relative γ. c Taken from the MCM v3.3.1 for general ambient scenarios.
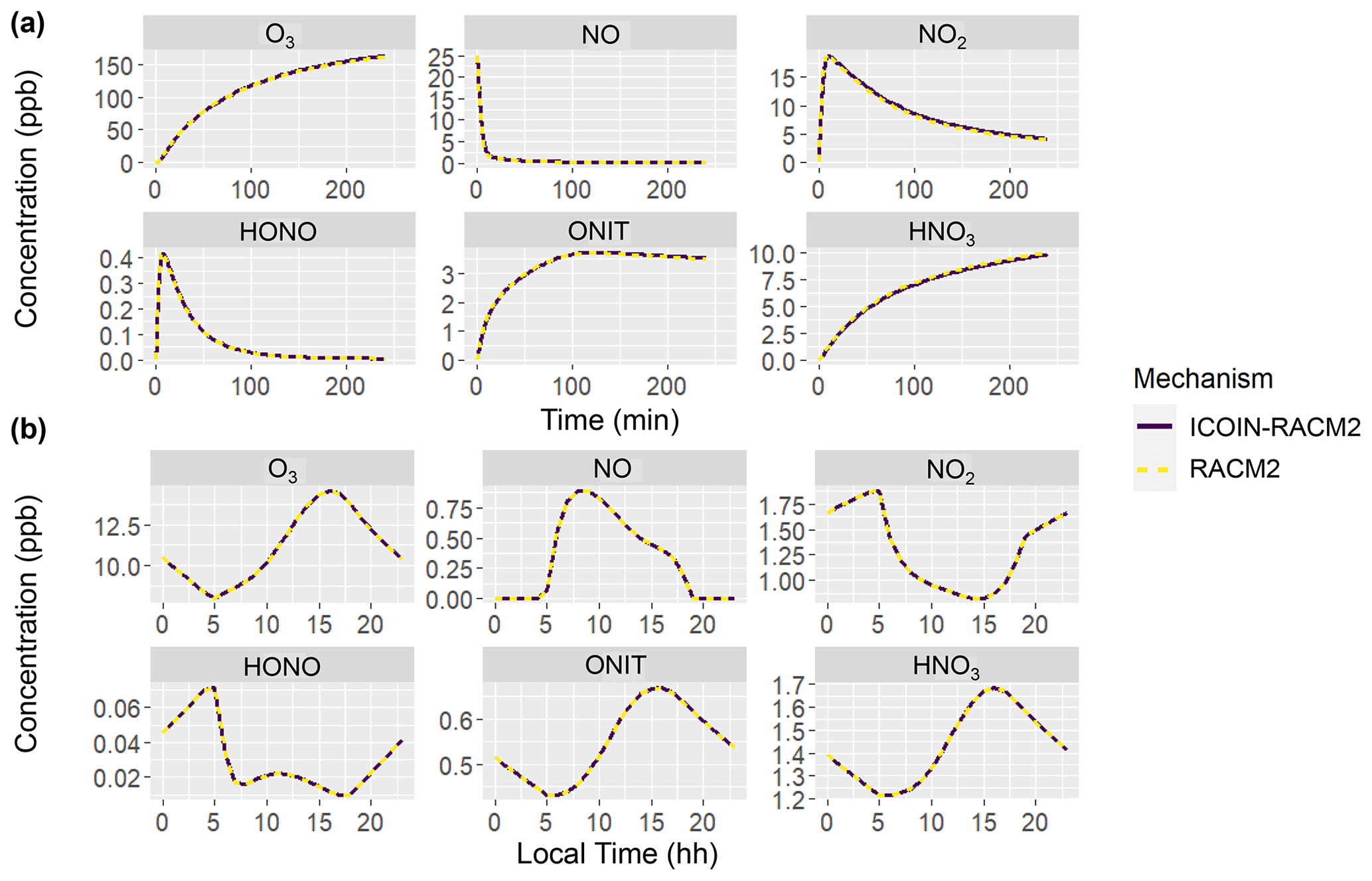
Figure 1Comparison of the RACM2 (dashed yellow line) and ICOIN-RACM2 (solid purple line) mechanisms for simulating concentrations of several molecules for (a) chamber experiment 3 and (b) diel cycle Case 19.
3.1 Mechanism evaluation
The efficacy of the isotope tagging methodology was assessed through a comparative analysis of molecule concentrations using both the RACM2 and ICOIN-RACM2 mechanisms. For molecules encompassing oxygen isotopologues explicitly considered in the ICOIN-RACM2 mechanism, the concentrations were derived by summing the isotopologue concentrations (Eq. 4).
where [X] refers to the concentration of a molecule with oxygen isotopologues, i refers to the unique oxygen isotopologues, and [Zi] refers to the concentration of the ith isotopologue. Both the RACM2 and ICOIN-RACM2 mechanisms simulated identical concentrations across both simulated scenarios: the hypothetical chamber experiments and the diurnal variation case study during the summer period (Fig. 1). This congruence in results aligns with expectations, as the isotope tagging approach implemented in the ICOIN-RACM2 is designed not to alter the chemical kinetics governing gas-phase reactions. Indeed, by definition, the presence of isotopes should remain inert with regard to chemical reactivity. This comparative analysis serves as a robust validation of the isotope tagging methodology's ability in simulating Δ17O values while maintaining the chemical reactivity stipulated by the RACM2 mechanism.
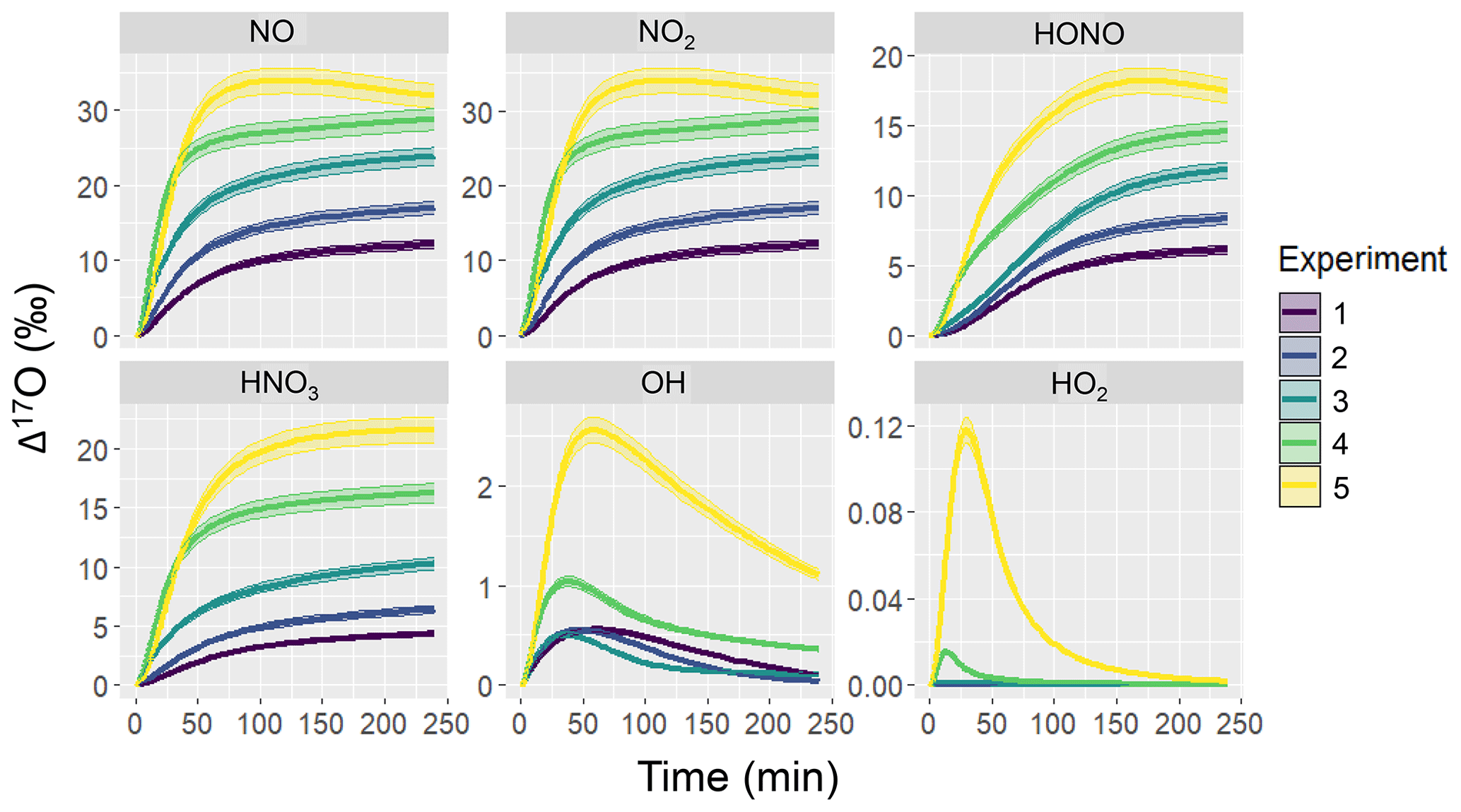
Figure 2Simulation of Δ17O for several NOy and Ox molecules including NO, NO2, HONO, HNO3, OH, and HO2 for the various hypothetical chamber experiments (color coded). The solid line represents the modeled Δ17O value and the shaded region corresponds to the propagated uncertainty associated with the chosen value of 39.3 ± 2 ‰. The experimental initial conditions are provided in Table 3, which include a starting NO concentration of 5, 12.5, 25, 62.5, and 125 ppb for experiments 1, 2, 3, 4, and 5, respectively.
3.2 Chamber simulations of NOx/α-pinene chemistry
The simulated Δ17O values derived from the hypothetical chamber simulations reveal significant temporal variations (Fig. 2). Overall, there were significant differences in Δ17O values across the considered molecules that increased in the sequence of HO2, OH, HNO3, HONO, and NO ≈ NO2. In the photochemical model simulations, the Δ17O(NO) ≈ Δ17O(NO2) due to both rapid NOx photochemical cycling and NO/NO2 isotope exchange. Among the considered NOy molecules, the initial Δ17O values start at 0 ‰ and subsequently rises due to the generation of O3 and subsequent propagation into the NOy components, which leads to heightened Δ17O values. The extent of Δ17O elevation was determined to be contingent upon the initial chamber conditions, becoming more pronounced with increasing initial NO to α-pinene ratios for Δ17O of NO, NO2, HONO, and HNO3. In contrast, Δ17O(HO2) was nearly negligible, aligning with common assumptions in other Δ(17O) models (Alexander et al., 2020, 2009; Michalski et al., 2003; Morin et al., 2011). Similarly, Δ17O(OH) generally maintained close proximity to 0 ‰, in line with typical assumptions in other Δ(17O) models (Alexander et al., 2020, 2009; Michalski et al., 2003; Morin et al., 2011); however, there were instances that deviated from this trend. Notably, higher Δ17O(OH) values were observed as the initial NOx relative to biogenic VOC (BVOC) concentrations increased. This occurrence can be attributed to the increased significance of oxygen isotope exchange between NO2 and OH for the higher initial NOx experimental conditions.
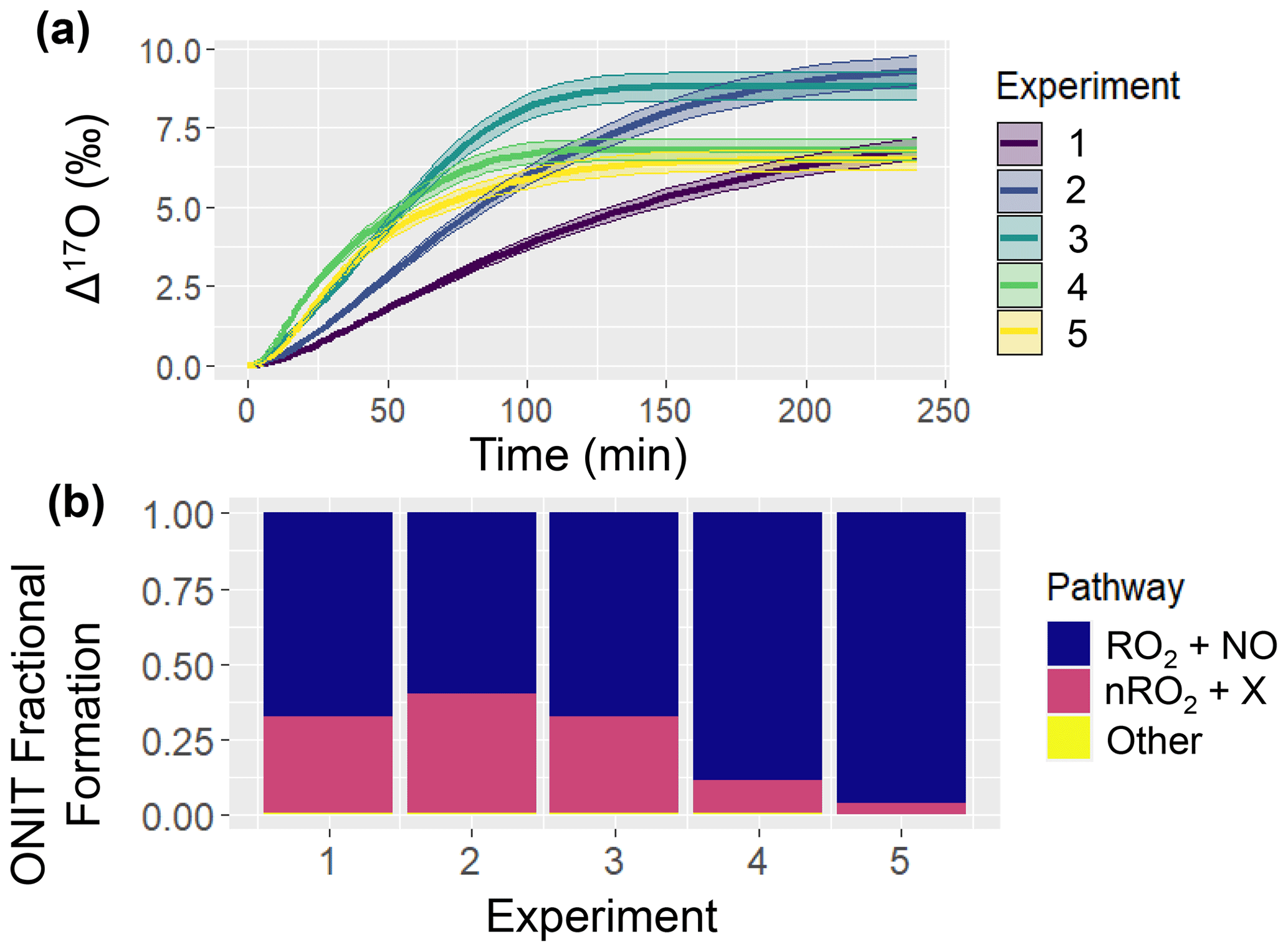
Figure 3Simulation of ONIT chemistry for the various considered chamber experiments including (a) Δ17O of ONIT and (b) ONIT fractional formation pathways. The solid line represents the modeled Δ17O value, and the shaded region corresponds to the propagated uncertainty associated with the chosen value of 39.3 ± 2 ‰ (a). The experimental initial conditions are provided in Table 3, which include a starting NO concentration of 5, 12.5, 25, 62.5, and 125 ppb for experiments 1, 2, 3, 4, and 5, respectively.
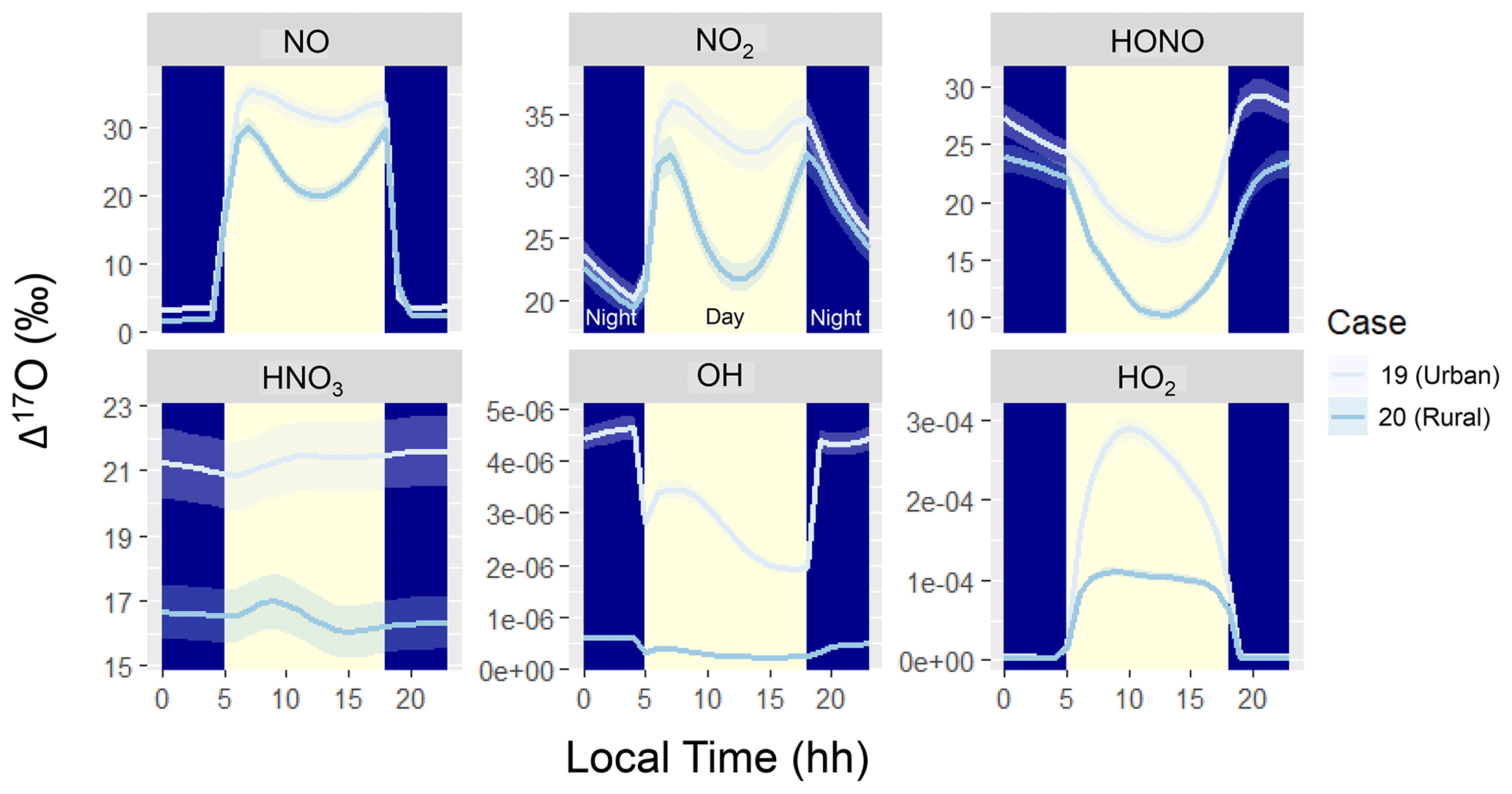
Figure 4Diel simulation of Δ17O for several NOy and Ox molecules including NO, NO2, HONO, HNO3, OH, and HO2 for the various hypothetical summertime cases (color coded). The solid line represents the modeled Δ17O value, and the shaded region corresponds to the propagated uncertainty associated with the chosen value of 39.3 ± 2‰. The shading corresponds to daytime (light yellow) and nighttime (dark blue) conditions.
An intriguing observation was that the simulated Δ17O(ONIT) values remained unaffected by the chamber's initial conditions (Fig. 3). This observation underscores the diverse ONIT formation pathways present in the experiments, encompassing a Δ17O(ONIT) low-end pathway involving the α-pinene peroxy radical (APIP; a type of RO2) + NO and a high-end pathway involving nitrooxy peroxy (nRO2) deriving from α-pinene + NO3 reactions (Table 1). We note that even though all of the simulated experiments were conducted under photochemical conditions, the model predicted some oxidation of α-pinene with NO3. The relative proportion of these two significant ONIT formation routes exhibited substantial variability across the various experiments (Fig. 3). Generally, a higher fractional formation of ONIT occurred through the Δ17O high-end member pathway of nRO2 + Y (where Y = HO2, NO, nRO2, ACO3, MO2) as the initial NO to α-pinene ratios were lower. Additionally, we note that the produced Δ17O(ONIT) value is a balance between the ONIT production pathway and the Δ17O(NO) (Table 1). For the lower initial [NOx]:[α-pinene] experiments, a lower Δ17O(NO) value was simulated. The balance between Δ17O(NO) and the pathway leading to ONIT production can explain the observation that Δ17O(ONIT) was insensitive to initial conditions.
The simulated Δ17O values from the chamber experiments highlight compelling dynamics. Primarily, there were substantial differences in Δ17O values arising from different formation pathways contributing to ONIT production. The uncertain nature of branching ratios and product yields for ONIT underscores the potential utility of comparing Δ17O(ONIT) observations with model simulations, aiding in the refinement of our understanding of ONIT yields originating from APIP + NO and API + NO3 reaction pathways. Furthermore, a significant divergence between HNO3 and ONIT in terms of Δ17O was observed, particularly with heightened initial NO to α-pinene concentrations. This divergence led to considerably higher simulated Δ17O(HNO3) than Δ17O(ONIT). This difference could potentially be utilized to help constrain the contribution of ONIT hydrolysis to HNO3 through a comparison of observed Δ17O(HNO3) and model-based predictions. Lastly, the model simulations underscore the potential of employing the ICOIN-RACM2 model to corroborate Δ17O values for OH and HO2 under diverse conditions (such as concentrations, chemical composition, relative humidity, and temperature), which are commonly assumed to be near 0 ‰.
3.3 Summertime diel simulations
The simulated Δ17O diel profiles indicate interesting patterns for the various considered molecules, including NO, NO2, HONO, HNO3, OH, and HO2 (Fig. 4). The Δ17O of NO, NO2, and HONO indicates a strong diurnal pattern for both considered summertime case studies. The simulated Δ17O of NO and NO2 indicates lower values during the nighttime and higher values during the daytime. This is due to the importance of nighttime NO emissions, such that the O atoms of NO and NO2 are not photochemically cycled. The simulated daytime profiles of Δ17O of NO and NO2 follow similar patterns, reflecting their fast photochemical cycling. Near sunrise, Δ17O of NO and NO2 reaches a peak due to photochemical cycling that primarily involves O3. As photolysis continues, there is a significant enhancement of peroxy radicals (), which slightly dilutes the Δ17O of NO and NO2. Near sunset, the peroxy radical concentrations decrease, and once again NO and NO2 predominantly photochemically cycle with O3. During the daytime, the simulated Δ17O(NO2) ≈ Δ17O(NO) due to the rapid NOx photochemical cycling. However, during the nighttime, Δ17O(NO2) was greater than Δ17O(NO) due to the role of nighttime NO emissions with an assumed Δ17O(NO) = 0 ‰. While NO and NO2 isotope exchange would lead to Δ17O(NO) = Δ17O(NO2), its role in influencing Δ17O depends on the concentrations of NO and NO2, as previously discussed for δ15N of NOx (Walters et al., 2016). In the diel model simulations, nighttime NO concentrations were less than 0.1 ppb (Fig. 1) due to its titration by O3. Under these conditions, the rate of NO/NO2 isotope exchange was slow relative to NO oxidation or the rate of NO primary emission, leading to a low nighttime Δ17O(NO) value for the simulation conditions of low nighttime NOx relative to O3 concentrations.
The predicted NO2 diurnal cycles of elevated Δ17O during the daytime and low Δ17O during the nights are generally consistent with summertime δ18O observations (which track with Δ17O) in West Lafayette, IN, US (Walters et al., 2018), and recent diel observations of Δ17O at Grenoble, FR, during the spring (Albertin et al., 2021). However, there are some slight differences in the daytime Δ17O(NO2) observations compared to the model simulations, in which the highest Δ17O(NO2) occurred for samples collected between 9 am–12 pm (Albertin et al., 2021). In comparison, the model indicated the highest Δ17O(NO2) around 6 to 8 am following the return of photolysis near sunrise. The observations indicate a subsequent daytime decay of Δ17O(NO2) (Albertin et al., 2021). The model also indicates a daytime decay in Δ17O(NO2) following the initial maximum Δ17O(NO2) that coincides with the onset of photolysis; however, the model expects an increase in Δ17O(NO2) in the late afternoon due to increased levels from the decrease in actinic flux. We do not intend to accurately simulate the previously reported Δ17O(NO2) values (Albertin et al., 2021). Some of the nuanced differences between the model simulation and observations of Δ17O are likely due to differences in meteorological conditions, as the model was simulated for summertime while the observations were from springtime and for a different latitude and longitude. Further, our model neglects transport and assumes a constant emission rate, which could influence the diel Δ17O(NO2) predictions. Nevertheless, the ICOIN-RACM2 mechanism appears to capture the general diurnal trend of Δ17O(NO2). We envision that future adaptation of the ICOIN-RACM2 mechanism into a chemical transport model would provide useful insight for constraining NOx photochemical cycling based on a comparison to field Δ17O(NO2) measurements.
The diurnal variation in Δ17O(HONO) exhibits an inverse pattern compared to NO and NO2, characterized by nocturnal maxima and daytime minima. This contrast arises from distinct formation pathways operating during daytime and nighttime. For our model simulations and conditions, HONO formation predominantly occurs via NO2 heterogeneous reactions during the night, giving rise to a high-Δ17O(HONO) end-member (Table 1). Conversely, daytime HONO production centers around the NO + OH pathway, leading to a low-Δ17O(HONO) end-member, which dilutes the Δ17O of the formed HONO relative to Δ17O of HONO and NO (Table 1). Notably, primary emissions could significantly contribute to HONO levels but were excluded from the hypothetical summertime scenarios (Stockwell et al., 1997). If primary HONO emissions were substantial, a lower Δ17O(HONO) during the night would be anticipated due to a lack of NOy photochemical cycling, assuming primary emissions carry a Δ17O(HONO) value of 0 ‰. Additionally, we note that the model, based on a gas-phase mechanism, does not include photolysis of pNO3, which could be an important source of HONO (Ye et al., 2016). For future interpretation of Δ17O(HONO) observations, photolysis of pNO3 and optimized NO2 heterogeneous reaction rates would need to be considered.
The Δ17O of HNO3, OH, and HO2 had little variation in their diel profiles. The Δ17O(HNO3) tended to converge to a value dependent on the oxidant conditions for Case 19 and Case 20. There was no significant simulated Δ17O(HNO3) diurnal variability due to the relatively longer HNO3 lifetime in the gas-phase mechanism relative to NO, NO2, and HONO, which rapidly undergo photochemical cycling. In the RACM2 mechanism, the significant chemical loss pathways for HNO3 are HNO3 + OH and HNO3 photolysis, which are relatively slow loss pathways, essentially “locking in” the Δ17O(HNO3) values. Thus, due to the relatively elevated HNO3 lifetime, the simulated Δ17O(HNO3) builds up toward a steady-state value. While the modeled diel Δ17O(HNO3) indicated no substantial diurnal variations, several field studies have indicated significant diurnal variability of Δ17O(pNO3) in polluted mega-cities (Zhang et al., 2022), as well as off the coast of California (Vicars et al., 2013). Commonly, Δ17O(HNO3) is thought to be equal to Δ17O(pNO3) due to the thermodynamic equilibrium between HNO3 and pNO3 in the fine aerosol mode (Alexander et al., 2009). However, recent data would suggest that Δ17O(HNO3) may not always be equal to Δ17O(pNO3) due to contributions of pNO3 in the coarse aerosol phase that may not achieve thermodynamic equilibrium with HNO3 (Kim et al., 2023). If we consider that the Δ17O(pNO3) diurnal variability should follow Δ17O(HNO3), the discrepancy between model and observations of diurnal variability would suggest that the lifetime of pNO3 in these previous studies must be shorter than predicted in our model for HNO3. Our model simulation was conducted using a gas-phase mechanism within a simple box model framework. Potentially important pNO3 loss processes not included in our model include pNO3 photolysis and wet or dry deposition. These processes should not alter the Δ17O of pNO3 but could reduce the lifetime of pNO3, leading to a significant diurnal variation in Δ17O. Additionally, our model simulation does not include transport or changes in boundary layer height and break up of the nocturnal boundary layer, which could also influence Δ17O diurnal variations of HNO3 and pNO3.
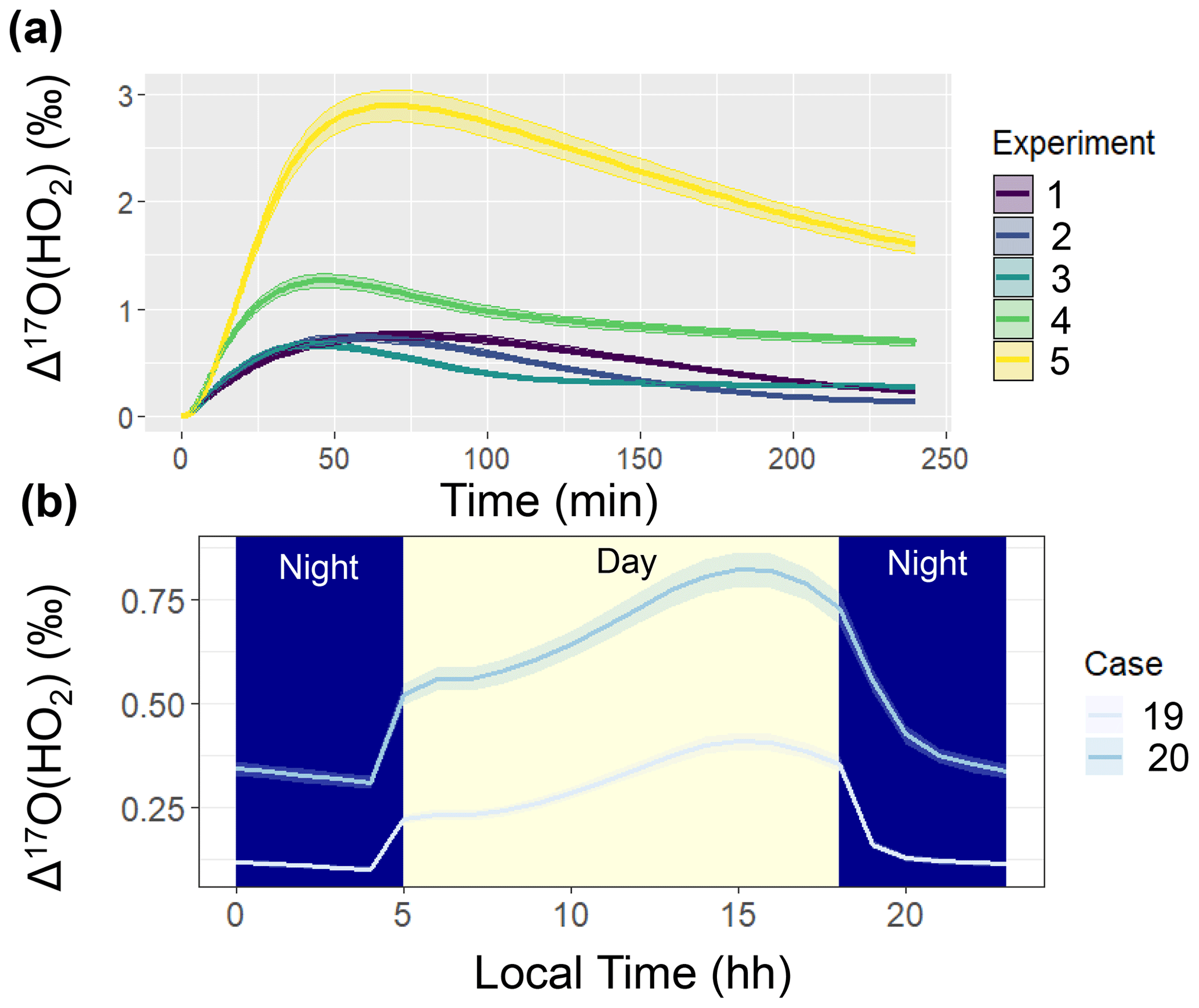
Figure 5Simulated Δ17O(HO2) when not considering oxygen isotope exchange between HO2 and O2 in the ICOIN-RACM2 mechanism (i.e., O-Exchange13 and O-Exchange14 in Table 2) for (a) the considered chamber simulations (color coded) and (b) the diel simulations for hypothetical summertime cases (color coded). The solid line represents the modeled Δ17O value, and the shaded region corresponds to the propagated uncertainty associated with the chosen value of 39.3 ± 2 ‰. The shading corresponds to daytime (light yellow) and nighttime (dark blue) conditions for the diel simulations (b).
The simulated Δ17O of OH and HO2 was near 0 ‰ for the entire simulation. We note that the simulated Δ17O(HO2) was lower than previous Δ17O(HO2) simulations (Morin et al., 2011), which tended to be between 1 ‰ to 2 ‰. This difference is because we have included oxygen isotope exchange reactions involving O2 and HO2 (Lyons, 2001) (i.e., O-Exchange13 and O-Exchange14 in Table 2) in the ICOIN-RACM2 mechanism, which rapidly remove Δ17O > 0 ‰ in the generated HO2. Without including this oxygen isotope exchange reaction, the ICOIN-RACM2 modeled Δ17O(HO2) predicts a non-zero Δ17O(HO2) that can be as high as 3 ‰ dependent on the model conditions (Fig. 5), consistent with previous model simulations (Morin et al., 2011). While the Δ17O(HO2) is expected to have a minor impact on the Δ17O of NOy species (Alexander et al., 2009), we should consider the importance of the role of oxygen isotope exchange between O2 and HO2 influencing Δ17O(HO2), as it will be an important source of Δ17O of H2O2, which is propagated into atmospheric sulfate (Savarino et al., 2000).
Comparing Case 19 and Case 20 reveals coherent diel Δ17O patterns. The primary disparity between these case studies lies in the higher Δ17O values exhibited by the considered NOy compounds in the urban setting of Case 19 compared to the rural backdrop of Case 20 (Fig. 4). The general divergence between the urban and rural simulated Δ17O stems from the interplay between O3 and . Urban conditions (i.e., Case 19) entail greater contributions from NOx photochemical cycling with O3 relative to the rural environment (i.e., Case 20). An important exception to the urban vs. rural trend was for Δ17O(HO2) when O2/HO2 exchange reactions were not considered in the model simulation (Fig. 5). From these simulations, the rural Δ17O(HO2) was slightly larger than the urban simulations, reflecting differences in the involvement of O3 in HO2 formation between these simulations. Overall, these hypothetical simulations highlight the prospect of intriguing Δ17O variations between urban and rural settings.
This study introduces a novel gas-phase mechanism, denoted as ICOIN-RACM2, which is built upon the RACM2 gas-phase chemical mechanism framework (Goliff et al., 2013). This mechanism is designed to explicitly model the Δ17O of NOy and Ox molecules based on quantitatively tracking the incorporation and propagation of the oxygen isotope anomaly derived from O3. Its application is demonstrated through box model simulations encompassing diverse hypothetical scenarios. These scenarios include chamber experiments focused on α-pinene and NO photochemical oxidation and the exploration of diurnal cycles in summertime chemistry.
These initial investigations serve as a fundamental step towards advancing our comprehension of NOy oxidation chemistry and its intricate pathways. Notably, the mechanism exhibits promising capabilities in simulating Δ17O values for multiple NOy and Ox species. This capacity holds considerable promise for refining our insights into aspects such as ONIT formation, branching ratios, and hydrolysis dynamics. Moreover, the ICOIN-RACM2 mechanism emerges as a valuable tool for various air-quality-related objectives. It could be an invaluable tool for the assessment of primary emission strengths for HONO and the probing of urban-to-rural gradients of atmospheric oxidation chemistry. In forthcoming endeavors, this newly devised model will be instrumental in direct comparisons with Δ17O observations arising from chamber experiments and investigations tied to air quality. As techniques for analyzing Δ17O in NOy molecules continue to advance, the model's utility is poised to expand.
An envisioned next step involves integrating the model into a broader 3-D atmospheric chemistry framework. This integration is anticipated to offer vital insights for evaluating the representation of oxidation chemistry across diverse landscapes. These endeavors have far-reaching implications, notably in fine-tuning our capacity to accurately model and predict atmospheric oxidation processes, thus enhancing our overall understanding of atmospheric oxidation capacity.
The developed mechanism, box model source codes, and the input and output files have been made publicly available at https://doi.org/10.5281/zenodo.10961373 (Walters, 2024).
The supplement related to this article is available online at: https://doi.org/10.5194/gmd-17-4673-2024-supplement.
WWW designed, tested, and evaluated the newly developed mechanism. MT and NLN provided critical insight into utilizing box models to simulate chamber experimental data. WWW and MGH secured funding for this work. WWW prepared the article with contributions from all co-authors.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
This research has been supported by NOAA's Climate Program Office's Atmospheric Chemistry, Carbon Cycle, and Climate program (NOAA AC4 NA18OAR4310118).
This paper was edited by Slimane Bekki and reviewed by two anonymous referees.
Albertin, S., Savarino, J., Bekki, S., Barbero, A., and Caillon, N.: Measurement report: Nitrogen isotopes (δ15N) and first quantification of oxygen isotope anomalies (Δ17O, δ18O) in atmospheric nitrogen dioxide, Atmos. Chem. Phys., 21, 10477–10497, https://doi.org/10.5194/acp-21-10477-2021, 2021. a, b, c, d, e, f
Alexander, B., Hastings, M. G., Allman, D. J., Dachs, J., Thornton, J. A., and Kunasek, S. A.: Quantifying atmospheric nitrate formation pathways based on a global model of the oxygen isotopic composition ((Δ17O) of atmospheric nitrate, Atmos. Chem. Phys., 9, 5043–5056, https://doi.org/10.5194/acp-9-5043-2009, 2009. a, b, c, d, e, f, g
Alexander, B., Sherwen, T., Holmes, C. D., Fisher, J. A., Chen, Q., Evans, M. J., and Kasibhatla, P.: Global inorganic nitrate production mechanisms: comparison of a global model with nitrate isotope observations, Atmos. Chem. Phys., 20, 3859–3877, https://doi.org/10.5194/acp-20-3859-2020, 2020. a, b, c, d, e, f, g, h
Barkan, E. and Luz, B.: High-precision measurements of and of O2 and ratio in air, Rapid Commun. Mass Sp., 17, 2809–2814, https://doi.org/10.1002/rcm.1267, 2003. a
Bhattacharya, S., Pandey, A., and Savarino, J.: Determination of intramolecular isotope distribution of ozone by oxidation reaction with silver metal, J. Geophys. Res.-Atmos., 113, D03303, https://doi.org/10.1029/2006JD008309, 2008. a
Blum, D. E., Walters, W. W., Eris, G., Takeuchi, M., Huey, L. G., Tanner, D., Xu, W., Rivera-Rios, J. C., Liu, F., Ng, N. L., and Hastings, M. G.: Collection of Nitrogen Dioxide for Nitrogen and Oxygen Isotope Determination–Laboratory and Environmental Chamber Evaluation, Anal. Chem, 95, 3371–3378, https://doi.org/10.1021/acs.analchem.2c04672, 2023. a, b
Browne, E. C. and Cohen, R. C.: Effects of biogenic nitrate chemistry on the NOx lifetime in remote continental regions, Atmos. Chem. Phys., 12, 11917–11932, https://doi.org/10.5194/acp-12-11917-2012, 2012. a
Chai, J., Dibb, J. E., Anderson, B. E., Bekker, C., Blum, D. E., Heim, E., Jordan, C. E., Joyce, E. E., Kaspari, J. H., Munro, H., Walters, W. W., and Hastings, M. G.: Isotopic evidence for dominant secondary production of HONO in near-ground wildfire plumes, Atmos. Chem. Phys., 21, 13077–13098, https://doi.org/10.5194/acp-21-13077-2021, 2021. a
Escorcia, E. N., Sjostedt, S. J., and Abbatt, J. P. D.: Kinetics of N2O5 Hydrolysis on Secondary Organic Aerosol and Mixed Ammonium Bisulfate–Secondary Organic Aerosol Particles, J. Phys. Chem. A, 114, 13113–13121, https://doi.org/10.1021/jp107721v, 2010. a
Fisher, J. A., Jacob, D. J., Travis, K. R., Kim, P. S., Marais, E. A., Chan Miller, C., Yu, K., Zhu, L., Yantosca, R. M., Sulprizio, M. P., Mao, J., Wennberg, P. O., Crounse, J. D., Teng, A. P., Nguyen, T. B., St. Clair, J. M., Cohen, R. C., Romer, P., Nault, B. A., Wooldridge, P. J., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Shepson, P. B., Xiong, F., Blake, D. R., Goldstein, A. H., Misztal, P. K., Hanisco, T. F., Wolfe, G. M., Ryerson, T. B., Wisthaler, A., and Mikoviny, T.: Organic nitrate chemistry and its implications for nitrogen budgets in an isoprene- and monoterpene-rich atmosphere: constraints from aircraft (SEAC4RS) and ground-based (SOAS) observations in the Southeast US, Atmos. Chem. Phys., 16, 5969–5991, https://doi.org/10.5194/acp-16-5969-2016, 2016. a
Galloway, J. N., Dentener, F. J., Capone, D. G., Boyer, E. W., Howarth, R. W., Seitzinger, S. P., Asner, G. P., Cleveland, C. C., Green, P. A., Holland, E. A., Karl, D. M., Michaels, A. F., Porter, J. H., Townsend, A. R., and Vöosmarty, C. J.: Nitrogen Cycles: Past, Present, and Future, Biogeochemistry, 70, 153–226, https://doi.org/10.1007/s10533-004-0370-0, 2004. a
Gao, Y. Q. and Marcus, R. A.: Strange and unconventional isotope effects in ozone formation, Science, 293, 259–263, https://doi.org/10.1126/science.1058528, 2001. a
Gen, M., Liang, Z., Zhang, R., Mabato, B. R. G., and Chan, C. K.: Particulate nitrate photolysis in the atmosphere, Environ. Sci. Atmos., 2, 111–127, https://doi.org/10.1039/D1EA00087J, 2022. a
Goliff, W. S., Stockwell, W. R., and Lawson, C. V.: The regional atmospheric chemistry mechanism, version 2, Atmos. Environ., 68, 174–185, https://doi.org/10.1016/j.atmosenv.2012.11.038, 2013. a, b, c, d, e
Hastings, M. G., Sigman, D. M., and Lipschultz, F.: Isotopic evidence for source changes of nitrate in rain at Bermuda, J. Geophys. Res.-Atmos., 108, 4790, https://doi.org/10.1029/2003JD003789, 2003. a
Holmes, C. D., Bertram, T. H., Confer, K. L., Graham, K. A., Ronan, A. C., Wirks, C. K., and Shah, V.: The Role of Clouds in the Tropospheric NOx Cycle: A New Modeling Approach for Cloud Chemistry and Its Global Implications, Geophys. Res. Lett., 46, 4980–4990, https://doi.org/10.1029/2019GL081990, 2019. a
Ishino, S., Hattori, S., Savarino, J., Jourdain, B., Preunkert, S., Legrand, M., Caillon, N., Barbero, A., Kuribayashi, K., and Yoshida, N.: Seasonal variations of triple oxygen isotopic compositions of atmospheric sulfate, nitrate, and ozone at Dumont d'Urville, coastal Antarctica, Atmos. Chem. Phys., 17, 3713–3727, https://doi.org/10.5194/acp-17-3713-2017, 2017. a, b, c
Janssen, C.: Intramolecular isotope distribution in heavy ozone (16O18O16O and 16O16O18O), J. Geophys. Res.-Atmos., 110, https://doi.org/10.1029/2004JD005479, 2005. a
Johnston, J. C. and Thiemens, M. H.: The isotopic composition of tropospheric ozone in three environments, J. Geophys. Res.-Atmos., 102, 25395–25404, https://doi.org/10.1029/97JD02075, 1997. a
Kim, H., Walters, W. W., Bekker, C., Murray, L. T., and Hastings, M. G.: Nitrate chemistry in the northeast US – Part 2: Oxygen isotopes reveal differences in particulate and gas-phase formation, Atmos. Chem. Phys., 23, 4203–4219, https://doi.org/10.5194/acp-23-4203-2023, 2023. a, b
Krankowsky, D., Lämmerzahl, P., and Mauersberger, K.: Isotopic measurements of stratospheric ozone, Geophys. Res. Lett., 27, 2593–2595, https://doi.org/10.1029/2000GL011812, 2000. a
Krankowsky, D., Lämmerzahl, P., Mauersberger, K., Janssen, C., Tuzson, B., and Röckmann, T.: Stratospheric ozone isotope fractionations derived from collected samples, J. Geophys. Res.-Atmos., 112, D08301, https://doi.org/10.1029/2006JD007855, 2007. a
Liu, Q., Schurter, L. M., Muller, C. E., Aloisio, S., Francisco, J. S., and Margerum, D. W.: Kinetics and mechanisms of aqueous ozone reactions with bromide, sulfite, hydrogen sulfite, iodide, and nitrite ions, Inorg. chem., 40, 4436–4442, https://doi.org/10.1021/ic000919j, 2001. a
Lyons, J. R.: Transfer of mass-independent fractionation in ozone to other oxygen-containing radicals in the atmosphere, Geophys. Res. Lett., 28, 3231–3234, https://doi.org/10.1029/2000GL012791, 2001. a, b, c, d
Marcus, R.: Mass-independent oxygen isotope fractionation in selected systems. Mechanistic considerations, Adv. Quantum Chem., 55, 5–19, https://doi.org/10.1016/S0065-3276(07)00202-X, 2008. a
Mauersberger, K., Lämmerzahl, P., and Krankowsky, D.: Stratospheric ozone isotope enrichments–revisited, Geophys. Res. Lett., 28, 3155–3158, https://doi.org/10.1029/2001GL013439, 2001. a
Michalski, G. and Bhattacharya, S.: The role of symmetry in the mass independent isotope effect in ozone, P. Natl. Acad. Sci. USA, 106, 5493–5496, https://doi.org/10.1073/pnas.0812755106, 2009. a
Michalski, G., Scott, Z., Kabiling, M., and Thiemens, M. H.: First measurements and modeling of Δ17O in atmospheric nitrate, Geophys. Res. Lett., 30, 1870, https://doi.org/10.1029/2003GL017015, 2003. a, b, c, d, e, f
Michalski, G., Bhattacharya, S. K., and Mase, D. F.: Oxygen isotope dynamics of atmospheric nitrate and its precursor molecules, in: Handbook of Environmental Isotope Geochemistry, edited by: Baskaran, M., Springer, New York, pp. 613–635, https://doi.org/10.1007/978-3-642-10637-8, 2012. a, b, c
Michalski, G., Bhattacharya, S. K., and Girsch, G.: NOx cycle and the tropospheric ozone isotope anomaly: an experimental investigation, Atmos. Chem. Phys., 14, 4935–4953, https://doi.org/10.5194/acp-14-4935-2014, 2014. a
Morin, S., Sander, R., and Savarino, J.: Simulation of the diurnal variations of the oxygen isotope anomaly (Δ17O) of reactive atmospheric species, Atmos. Chem. Phys., 11, 3653–3671, https://doi.org/10.5194/acp-11-3653-2011, 2011. a, b, c, d, e, f, g
Morton, J., Barnes, J., Schueler, B., and Mauersberger, K.: Laboratory studies of heavy ozone, J. Geosphys. Res.-Atmos., 95, 901–907, https://doi.org/10.1029/JD095iD01p00901, 1990. a
Newsome, B. and Evans, M.: Impact of uncertainties in inorganic chemical rate constants on tropospheric composition and ozone radiative forcing, Atmos. Chem. Phys., 17, 14333–14352, https://doi.org/10.5194/acp-17-14333-2017, 2017. a
Ng, N. L., Brown, S. S., Archibald, A. T., Atlas, E., Cohen, R. C., Crowley, J. N., Day, D. A., Donahue, N. M., Fry, J. L., Fuchs, H., Griffin, R. J., Guzman, M. I., Herrmann, H., Hodzic, A., Iinuma, Y., Jimenez, J. L., Kiendler-Scharr, A., Lee, B. H., Luecken, D. J., Mao, J., McLaren, R., Mutzel, A., Osthoff, H. D., Ouyang, B., Picquet-Varrault, B., Platt, U., Pye, H. O. T., Rudich, Y., Schwantes, R. H., Shiraiwa, M., Stutz, J., Thornton, J. A., Tilgner, A., Williams, B. J., and Zaveri, R. A.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 17, 2103–2162, https://doi.org/10.5194/acp-17-2103-2017, 2017. a
Pinder, R. W., Davidson, E. A., Goodale, C. L., Greaver, T. L., Herrick, J. D., and Liu, L.: Climate change impacts of US reactive nitrogen, P. Natl. Acad. Sci. USA, 109, 7671–7675, https://doi.org/10.1073/pnas.1114243109, 2012. a
Prinn, R. G.: The cleansing capacity of the atmosphere, Annu. Rev. Env. Resour., 28, 29–57, https://doi.org/10.1146/annurev.energy.28.011503.163425, 2003. a
Röckmann, T., Brenninkmeijer, C., Saueressig, G., Bergamaschi, P., Crowley, J., Fischer, H., and Crutzen, P.: Mass-independent oxygen isotope fractionation in atmospheric CO as a result of the reaction CO + OH, Science, 281, 544–546, https://doi.org/10.1126/science.281.5376.544, 1998. a, b
Romer, P. S., Duffey, K. C., Wooldridge, P. J., Allen, H. M., Ayres, B. R., Brown, S. S., Brune, W. H., Crounse, J. D., de Gouw, J., Draper, D. C., Feiner, P. A., Fry, J. L., Goldstein, A. H., Koss, A., Misztal, P. K., Nguyen, T. B., Olson, K., Teng, A. P., Wennberg, P. O., Wild, R. J., Zhang, L., and Cohen, R. C.: The lifetime of nitrogen oxides in an isoprene-dominated forest, Atmos. Chem. Phys., 16, 7623–7637, https://doi.org/10.5194/acp-16-7623-2016, 2016. a
Savarino, J., Lee, C., and Thiemens, M.: Laboratory oxygen isotopic study of sulfur (IV) oxidation: Origin of the mass-independent oxygen isotopic anomaly in atmospheric sulfates and sulfate mineral deposits on Earth (Paper 2000JD900456), J. Geophys. Res.-Atmos., 105, 29–079, https://doi.org/10.1029/2000JD900456, 2000. a
Savarino, J., Vicars, W. C., Legrand, M., Preunkert, S., Jourdain, B., Frey, M. M., Kukui, A., Caillon, N., and Gil Roca, J.: Oxygen isotope mass balance of atmospheric nitrate at Dome C, East Antarctica, during the OPALE campaign, Atmos. Chem. Phys., 16, 2659–2673, https://doi.org/10.5194/acp-16-2659-2016, 2016. a
Stockwell, W. R., Kirchner, F., Kuhn, M., and Seefeld, S.: A new mechanism for regional atmospheric chemistry modeling, J. Geophys. Res.-Atmos., 102, 25847–25879, https://doi.org/10.1029/97JD00849, 1997. a, b, c, d, e
Takeuchi, M. and Ng, N. L.: Chemical composition and hydrolysis of organic nitrate aerosol formed from hydroxyl and nitrate radical oxidation of α-pinene and β-pinene, Atmos. Chem. Phys., 19, 12749–12766, https://doi.org/10.5194/acp-19-12749-2019, 2019. a, b
Thiemens, M. H. and Jackson, T.: Pressure dependency for heavy isotope enhancement in ozone formation, Geophys. Res. Lett., 17, 717–719, https://doi.org/10.1029/GL017i006p00717, 1990. a
Velivetskaya, T. A., Ignatiev, A. V., Budnitskiy, S. Y., Yakovenko, V. V., and Vysotskiy, S. V.: Mass-independent fractionation of oxygen isotopes during H2O2 formation by gas-phase discharge from water vapour, Geochim. Cosmochim. Ac., 193, 54–65, https://doi.org/10.1016/j.gca.2016.08.008, 2016. a, b, c
Vicars, W. C., Morin, S., Savarino, J., Wagner, N. L., Erbland, J., Vince, E., Martins, J. M. F., Lerner, B. M., Quinn, P. K., Coffman, D. J., Williams, E. J., and Brown, S. S.: Spatial and diurnal variability in reactive nitrogen oxide chemistry as reflected in the isotopic composition of atmospheric nitrate: Results from the CalNex 2010 field study, Geophys. Res. Atmos., 118, 10–567, https://doi.org/10.1002/jgrd.50680, 2013. a
Vicars, W. C. and Savarino, J.: Quantitative constraints on the 17O-excess (Δ17O) signature of surface ozone: Ambient measurements from 50° N to 50° S using the nitrite-coated filter technique, Geochim. Cosmochim. Ac., 135, 270–287, https://doi.org/10.1016/j.gca.2014.03.023, 2014. a, b, c
Vicars, W. C., Bhattacharya, S., Erbland, J., and Savarino, J.: Measurement of the 17O-excess (Δ17O) of tropospheric ozone using a nitrite-coated filter, Rapid Commun. Mass Sp., 26, 1219–1231, https://doi.org/10.1002/rcm.6218, 2012. a
Walters, W.: ICOIN_RACM2 (v1.3), Zenodo [code and data set], https://doi.org/10.5281/zenodo.10961373, 2024. a
Walters, W. W. and Michalski, G.: Ab initio study of nitrogen and position-specific oxygen kinetic isotope effects in the NO + O3 reaction, J. Chem. Phys., 145, 224311, https://doi.org/10.1063/1.4968562, 2016. a
Walters, W. W., Simonini, D. S., and Michalski, G.: Nitrogen isotope exchange between NO and NO2 and its implications for δ15N variations in tropospheric NOx and atmospheric nitrate, Geophys. Res. Lett., 43, 440–448, https://doi.org/10.1002/2015GL066438, 2016. a
Walters, W. W., Fang, H., and Michalski, G.: Summertime diurnal variations in the isotopic composition of atmospheric nitrogen dioxide at a small midwestern United States city, Atmos Environ, 179, 1–11, https://doi.org/10.1016/j.atmosenv.2018.01.047, 2018. a, b
Walters, W. W., Michalski, G., Böhlke, J. K., Alexander, B., Savarino, J., and Thiemens, M. H.: Assessing the Seasonal Dynamics of Nitrate and Sulfate Aerosols at the South Pole Utilizing Stable Isotopes, J. Geophys. Res.-Atmos., 124, 8161–8177, https://doi.org/10.1029/2019JD030517, 2019. a
Wang, Y., Piletic, I. R., Takeuchi, M., Xu, T., France, S., and Ng, N. L.: Synthesis and hydrolysis of atmospherically relevant monoterpene-derived organic nitrates, Environ. Sci. Technol., 55, 14595–14606, https://doi.org/10.1021/acs.est.1c05310, 2021. a
Wang, Y., Takeuchi, M., Wang, S., Nizkorodov, S. A., France, S., Eris, G., and Ng, N. L.: Photolysis of gas-phase atmospherically relevant monoterpene-derived organic nitrates, J. Phys. Chem. A, 127, 987–999, https://doi.org/10.1021/acs.jpca.2c04307, 2023. a
Wolfe, G. M., Marvin, M. R., Roberts, S. J., Travis, K. R., and Liao, J.: The Framework for 0-D Atmospheric Modeling (F0AM) v3.1, Geosci. Model Dev., 9, 3309–3319, https://doi.org/10.5194/gmd-9-3309-2016, 2016. a, b, c
Ye, C., Zhou, X., Pu, D., Stutz, J., Festa, J., Spolaor, M., Tsai, C., Cantrell, C., Mauldin, R. L., Campos, T., Weinheimer, A., Hornbrook, R. S., Apel, E. C., Guenther, A., Kaser, L., Yuan, B., Karl, T., Haggerty, J., Hall, S., Ullmann, K., Smith, J. N., Ortega, J., and Knote, C.: Rapid cycling of reactive nitrogen in the marine boundary layer, Nature, 532, 489–491, https://doi.org/10.1038/nature17195, 2016. a
Zare, A., Romer, P. S., Nguyen, T., Keutsch, F. N., Skog, K., and Cohen, R. C.: A comprehensive organic nitrate chemistry: insights into the lifetime of atmospheric organic nitrates, Atmos. Chem. Phys., 18, 15419–15436, https://doi.org/10.5194/acp-18-15419-2018, 2018. a, b
Zhang, Y.-L., Zhang, W., Fan, M.-Y., Li, J., Fang, H., Cao, F., Lin, Y.-C., Wilkins, B. P., Liu, X., Bao, M., Hong, Y., and Michalski, G.: A diurnal story of in urban Nanjing and its implication for nitrate aerosol formation, npj Clim. Atmos. Sci., 5, 50, https://doi.org/10.1038/s41612-022-00273-3, 2022. a