the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 21 Dec 2020
| 21 Dec 2020
GenChem v1.0 – a chemical pre-processing and testing system for atmospheric modelling
Robert Bergström
Alan Briolat
Hannah Imhof
John Johansson
Michael Priestley
Alvaro Valdebenito
This paper outlines the structure and usage of the GenChem system, which includes a chemical pre-processor GenChem.py) and a simple box model (boxChem). GenChem provides scripts and input files for converting chemical equations into differential form for use in atmospheric chemical transport models (CTMs) and/or the boxChem system. Although GenChem is primarily intended for users of the Meteorological Synthesizing Centre – West of the European Monitoring and Evaluation Programme (EMEP MSC-W) CTM and related systems, boxChem can be run as a stand-alone chemical solver, enabling for example easy testing of chemical mechanisms against each other. This paper presents an outline of the usage of the GenChem system, explaining input and output files, and presents some examples of usage.
The code needed to run GenChem is released as open-source code under the GNU license.
- Article
(1032 KB) - Full-text XML
-
Supplement
(233 KB) - BibTeX
- EndNote





Atmospheric chemical transport models (CTMs) – which simulate the emissions, transport, chemistry and loss processes of pollutants – are essential tools for understanding air quality and for assisting governments in setting environmental goals and emissions targets. Such CTMs are typically advanced three-dimensional models with perhaps a million grid cells. The models account for transport (advection and dispersion) between the cells, and within each cell the chemistry of the atmosphere is simulated, usually with a “condensed” chemical mechanism (see below) and time steps ranging from seconds to minutes.
An important CTM in terms of policy is that used by the Meteorological Synthesising Centre – West of the European Monitoring and Evaluation Programme (EMEP MSC-W). The EMEP MSC-W CTM (hereafter EMEP CTM), described in detail in Simpson et al. (2012) and subsequent articles and EMEP reports (e.g. Stadtler et al., 2018; Simpson et al., 2020a, and references therein), is a three-dimensional Eulerian model whose main aim is to support governments in their efforts to design effective emissions control strategies. The EMEP CTM has been available as open-source code (http://www.emep.int, last access: 2 December 2020) since 2008, and it has since been run by several institutes across Europe (e.g. Solberg et al., 2008; Jeričevič et al., 2010; Karl et al., 2014; Omstedt et al., 2015; Vieno et al., 2016; Ots et al., 2018).
As with most CTM systems, the EMEP CTM code does not directly read chemical equations but rather requires the production and loss terms of each species to be specified, in a differential form suitable for numerical integration. In order to convert between chemical equations and the numerical form, a chemical pre-processor is used, together with support software, which together comprise the “GenChem” system.
In addition to the three-dimensional EMEP CTM, a 1-D model system, the Ecosystem Surface Exchange model (ESX; Simpson and Tuovinen, 2014), is being developed as a complementary system. ESX allows for the investigation of for example chemistry and deposition processes within the lowest tens of metres of the atmosphere (similar in concept to e.g. Makar et al., 1999; Ashworth et al., 2015). The most recent version of the ESX model also allows for Lagrangian trajectory simulations, which will enable the exploration of detailed chemical analysis as air masses traverse perhaps hundreds of kilometres (similar to e.g. Hertel et al., 1995; Vieno et al., 2010; Lowe et al., 2011; Murphy et al., 2011). ESX makes use of many components of the EMEP CTM, including many routines for, for example, radiation, emissions and the GenChem system.
The most-well-known chemical pre-processor is probably KPP (Kinetic PreProcessor; Damian et al., 2002; Sandu and Sander, 2006), which is used in a number of CTMs (e.g. Ashworth et al., 2015; Eller et al., 2009; Lowe et al., 2009; Langner et al., 1998; Squire et al., 2015; Stroud et al., 2016). KPP is more flexible than GenChem, with for example a range of different chemical solvers available and the capability to output as Fortran, C or MATLAB code. GenChem does not aim to compete with KPP in these regards, and in future KPP (or some descendent) may well replace GenChem in the EMEP CTM system also, but for the purposes of the EMEP CTM, GenChem does have a number of useful features:
-
The GenChem code is tailor-made to produce Fortran which can be directly included in the EMEP CTM and ESX systems.
-
The integrated boxChem system allows both direct testing of code prepared for the EMEP CTM and ESX model, and side-by-side comparison of chemical schemes.
-
The Fortran code produced is more human-readable than with other processors such as KPP, with for example the gas HNO3 being represented by the Fortran integer named “HNO3” instead of by some numeric or abstract variable representation. Similarly, equations in the code produced by GenChem are readily understood, as illustrated in Fig. 1.
-
The code also establishes dry- and wet-deposition mapping of the chemical species, as well as a number of other characteristics (which can be readably extended through the “Groups” system; see Sect. 6), such as volatility of organic aerosols or extinction coefficients for aerosol species.
-
The numerical solver, TWOSTEP (described below), is extremely efficient for 3-D chemical transport models and is thus very apt for the EMEP CTM or for running complex chemical mechanisms such as the Master Chemical Mechanism in boxChem or ESX (Sect. 5).
-
GenChem has a flexible system that can either calculate molecular weights from chemical formulas or use user-provided values.
The original GenChem system was written in Perl in the 1990s for earlier EMEP CTM versions (e.g. Simpson, 1995; Simpson et al., 2012) but was converted to a Python (2.7) script in 2014. The current structure of the GenChem system – now based entirely on Python 3, including boxChem, improvements in GenChem.py and various scripts – was developed between 2016 and 2020.

Figure 1Example equations from the output file, CM_Reactions1.inc, for the model species GLYOX (glyoxal). “P” gives the production terms, and “L” gives the loss terms, with the last line giving the TWOSTEP solution for this species. See Sect. 7.5.
The numerical solver used for the chemical equations in the EMEP CTM is the so-called TWOSTEP scheme (Verwer, 1994; Verwer et al., 1996; Verwer and Simpson, 1995). Sandu et al. (1997) commented that TWOSTEP was an excellent candidate for very large tropospheric gas-phase problems with very small operator split steps. The main limitation noted by Sandu et al. (1997) was that TWOSTEP is not suitable for aqueous-phase problems, but those are not explicitly treated within the gas-phase mechanisms considered here. It is not the purpose of this article to give details of the TWOSTEP scheme, except to note the simple formulation which results from its use. For example, Fig. 1 illustrates one of the major outputs of GenChem: example code (for the species glyoxal) from the GenChem output file CM_Reactions1.inc. This code is very easy to read, with first the production term (P), which includes time-varying rate coefficients (rct terms), photolysis rates (rcphot) and emissions (rcemis); then the chemical loss terms (L); and finally the Gauss–Seidel TWOSTEP solution for the change in concentrations over time step dt2. These terms are discussed further in Sects. 6.3 and 7.5.
Although primarily intended for users of the EMEP CTM, the GenChem system can also be run as a stand-alone chemical solver using the provided “boxChem” driver, enabling for example easy testing of chemical mechanisms against each other. boxChem also provides a useful learning tool for general GenChem usage. This paper outlines the structure of the GenChem system, including boxChem usage and preparation of EMEP-ready model files.
This paper is mainly intended as a complement to the user guide and code provided with GenChem, but we aim to provide here some more discussion of the background and benefits of the approaches chosen. Section 2 focuses on the installation and code structure of the GenChem system. Section 3 illustrates the steps needed to set up and run the boxChem simulations, including plotting commands. This allow users to get a quick start on the GenChem system, i.e. to actually run and compare chemical schemes. Section 4 explains how to create and transfer files to the EMEP CTM system. Section 5 explains the many possible options associated with the “base” and “extra” chemical mechanisms. Section 6 explains how to define the chemical mechanisms: detailing the input files which contain chemical species information and reaction mechanisms. Section 7 documents the output files of GenChem, which consist mainly of Fortran code needed for boxChem and EMEP CTM runs. Finally, Sect. 8 (Conclusions) discusses some ideas for future development of the GenChem system. In this paper we use bold-italic font to denote script and file names, bold font for directory names, and typewriter font for extracts of files. Variable names are simply denoted with default fonts.
The code needed to run the GenChem system is released as open-source code under the GNU license (https://github.com/metno/genchem, last access: 2 December 2020), with the user guide provided at https://genchem.readthedocs.io (last access: 2 December 2020). GenChem has been developed and tested in a Linux environment, mainly Xubuntu 16.04–19.10, but it has also been tested on Windows systems via a virtual environment. For those familiar with the Docker system (https://www.docker.com/products/docker-desktop, last access: 2 December 2020), a Dockerfile and Dockerfile_README are provided to enable consistent installation on Windows PCs. See the user guide for further details.
GenChem is designed to work with modern Fortran compilers (tested with GNU Fortran (GFortran) and Intel f95), together with Python 3 (≥3.5). As in the EMEP CTM, double precision is enforced by compiler options (e.g. -r8 for ifort) rather than through explicit Fortran “double precision” or “selected_real_kind” coding. This is partly for aesthetic reasons (we prefer numbers typed as 1.2 rather than 1.2_wp) and partly for simplicity. Use of these flags ensures that all variables and constants are automatically elevated to the required precision. Failure to compile with these options will result in an error message. The testing for this release has mainly been done with the freely available GNU Fortran version 5.
After unpacking the GenChem directory structure should look like Fig. 2. The chem/base_mechanisms directory contains the main gas-phase mechanisms which we will use (e.g. EmChem19a). These are almost always supplemented by additional smaller mechanisms (e.g. for aerosol formation or biogenic emissions) which are contained in the chem/extra_mechanisms directory; see Sect. 5 for more details. The scripts GenChem.py and do.GenChem reside within the chem/scripts directory, but we will usually make use of these from the box directory as explained in Sect. 3. The main technical documentation of this system is provided at https://genchem.readthedocs.io (last access: 2 December 2020) as noted above, but various markdown-format README.md files are also located throughout this structure. For example each chemical mechanism should have a README.md file to summarise the mechanism and any comments.
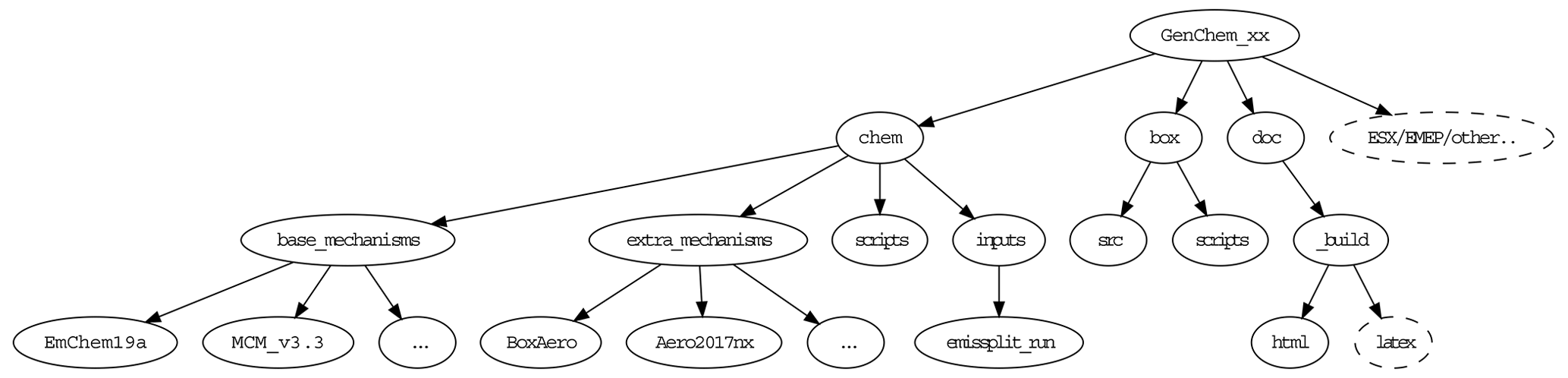
Figure 2Directory structure of GenChem's chem and box directories. The dashed “ESX/EMEP/other” directory indicates the possible placement of future EMEP, ESX or other model directories in this system.
The boxChem system is an integral part of the GenChem system. boxChem provides a way of testing GenChem implementations and is indeed strongly recommended as the main method of preparing EMEP CTM codes. As a stand-alone model, boxChem is also a valuable way to compare results from different chemical mechanisms.
3.1 Initial setup of boxChem
boxChem, and indeed all GenChem work, is usually done from a temporary “work” directory of the box system, for example from tmp_work. This is created from the box location of
- Example 3.1
-
./scripts/box_setup.sh tmp_work.
This will create the working directory and copy all the files needed for boxChem into it. Once it is set up, the user is ready to build and run some chemical schemes. With the example of EmChem19a, and now from our tmp_work directory, the simplest next step is in principle to run
- Example 3.2
-
./do.GenChem -b EmChem19a.
This will run the GenChem.py script on the base mechanism (-b flag) EmChem19a, and generate CM and CMX files, but it will not attempt to compile or run boxChem. However, a far more useful approach is to run do.testChems, for example
- Example 3.3
-
./do.testChems EmChem19a.
Running do.testChems will run GenChem.py on the EmChem19a scheme (also adding a few extra_mechanism files as discussed in Sect. 5) and copy all necessary CM files and the configuration file config_box.nml to the user's work directory. The script compiles boxChem and then runs the resulting box-model code. Input variables needed by boxChem (e.g. meteorology, emissions and boundary conditions) are set in config_box.nml (also copied by do.testChems).
Results of the boxChem run will appear as three files in the output directory as given in Table 2. The main result file uses a simple comma-separated format and is readable with for example LibreOffice. Plot scripts are also available for easy visualisation and comparison of these CSV (comma-separate values) results (Sect. 3.3).
Table 1Input files to the GenChem system and output Fortran files.
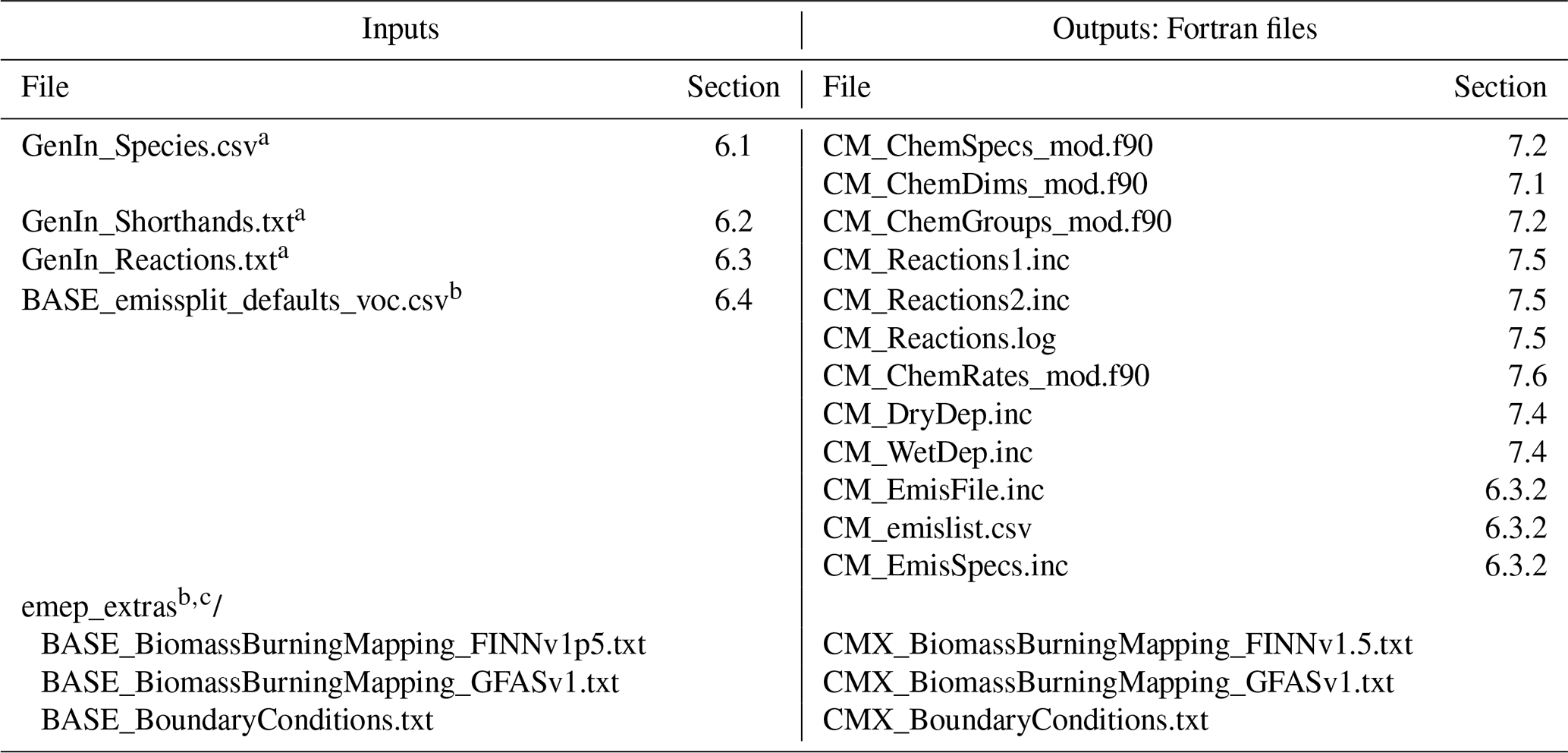
a These GenIn files are generated from one or more equivalent files; see Sects. 6.1–6.3. b For the input emissplit_defaults_voc.csv file and emep_extras the “BASE” string is replaced by the name of the base chemical mechanism, e.g. EmChem19a. c The files in emep_extras are for the EMEP CTM rather than boxChem usage. If intended for the EMEP CTM, then appropriate Fortran code is required. If for boxChem, only dummy files are provided. These files are essential only for the base mechanisms. See the README_emep_extras.md files in the sub-directory.
Table 2Outputsa for a boxChem run achieved via “do.testChems EmChem19a”.
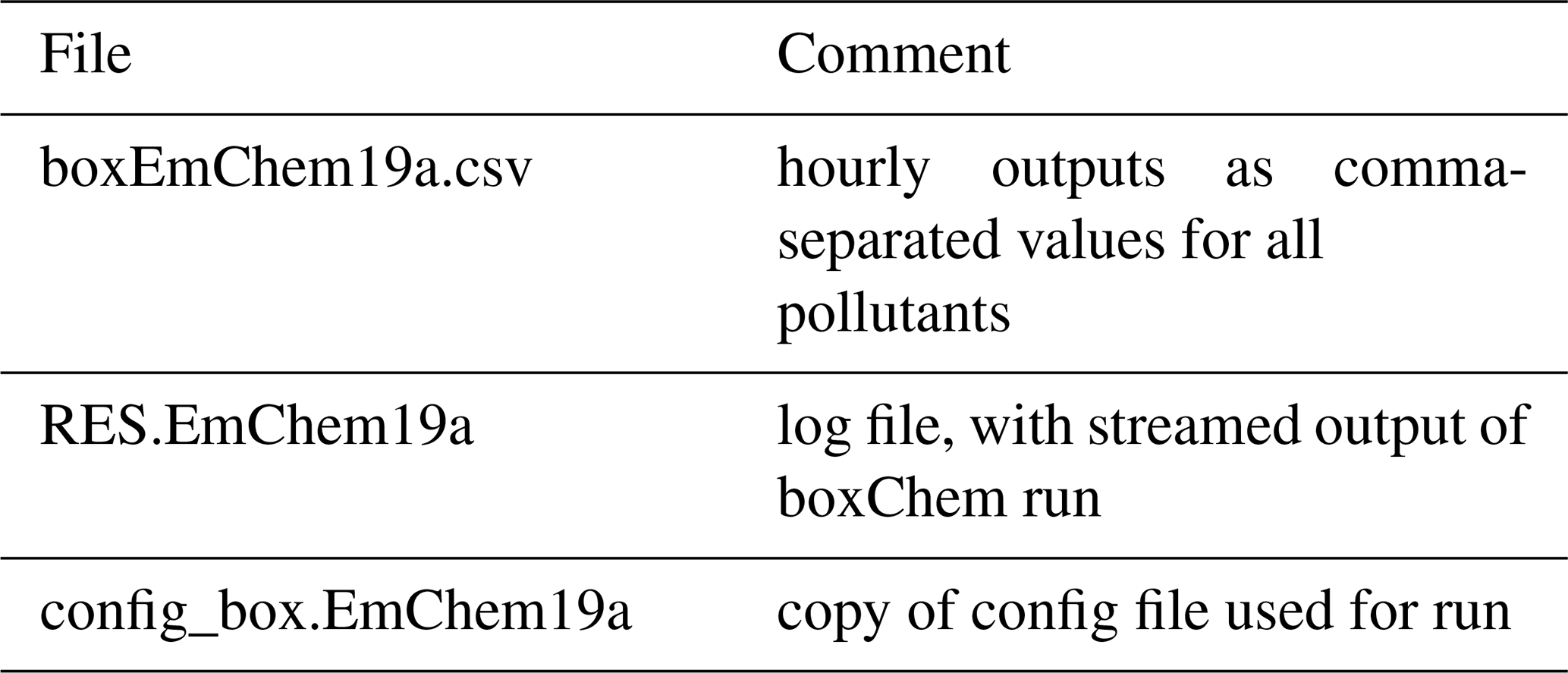
a Files are produced in the OUTPUTS directory by default and can be modified in do.testChems script.
The “CM_” and “CMX_” Fortran files produced by this process are saved in directories, for example here in the directory “ZCMBOX_EmChem19a”. Now, if one wants to compare several schemes, one can do for example
- Example 3.4
-
./do.testChems EmChem19a CB6r2Em CRIv2R5Em.
This would produce three output .csv files, which again are easily plotted against each other (see Sect. 3.3).
3.2 Controlling boxChem inputs and outputs: config_box.nml
The namelist input file config_box.nml allows the user to control many aspects of the boxChem model run. This file specifies the start and end time as well as the time step (dt) to be used. The concentration of the fixed species “M” and “H2O” (concentrations of air and water molecules; see Sect. 6.2) and initial concentrations of all species are set in config_box.nml. M and H2O can be set either directly in molecules per cubic centimetre or by defining the pressure and relative humidity, respectively. Variables such as temperature, relative humidity or anthropogenic emissions are also set in config_box.nml and stay constant over the simulation time. Photolysis rates, however, change every time step according to the Sun's zenith angle. Biogenic emissions may also be modified by zenith angle if the simple “SUN” variable is used (see Sect. 6.3.2).
By default, boxChem uses the set of emission rates as specified by
variables set in config_box.nml, currently set with the lines beginning
emis_kgm2day = 'nox', 18.3 ! NOx, kg/m2/day,
with “voc” emissions set on the next line as 15.4 kg m−2 d−1 (see Sect. 7.7). These emissions are converted by boxChem to instantaneous production rates in molecules per cubic centimetre per second, accounting for molecular masses; emissions speciation (e.g. NOx as NO and NO2); and the mixing height, Hmix (also set in config_box.nml). Such emission rates can be modified by the user, or indeed all emissions can be set to 0 if the variable use_emis is set to “F” (false). boxChem makes use of default speciation of compound emissions such as NOx or VOC – see Sects. 6.4 and S2.3 in the Supplement for more information on these splits and how they can be changed.
The “OutSpecs_list” variable in config_box.nml specifies which pollutants are required in the output file, though by default it is set to “all”. This output file, for example box_dt30s.csv (where 30 s is the “external” time step used), is generated in the outputs directory (e.g box/tmp_work/OUTPUTS) and gives hourly values for each species specified in OutSpecs_list, along with appropriate units. For gaseous species we use parts per billion or molecules per cubic centimetre, and for particulate matter (PM) micrograms per cubic metre.
The choice of time step is discussed in the Supplement. See the comments in config_box.nml for further details about boxChem and config_box.nml setup and usage.
3.3 Plotting boxChem outputs
The Python/matplotlib script boxplots.py (found in the box/scripts directory) can plot either individual or multiple species produced by boxChem, and for one or several output files. For example, if one has run do.testChems on, say, two chemical schemes, the results are easily plotted from the box/tmp_work/OUTPUTS directory:
- Example 3.5
-
../../scripts/boxplots.py -h,
where -h triggers a help message, -v gives the compound to be plotted, and -p produces a PNG graphics file as well as screen output. Using “ALL” or “DEF” with -v results in all or many common species being plotted at once (-p is assumed in this case). For example, Fig. 3 shows a comparison of three schemes produced with this script.
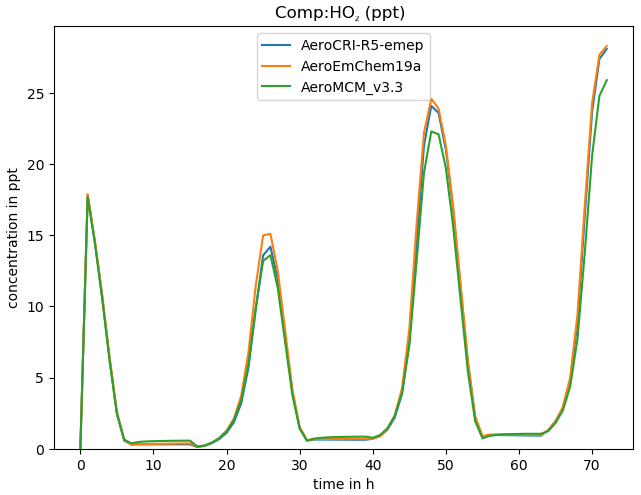
Figure 3Example of comparison of three chemical schemes, produced for HO2 with the boxplots.py script.
Another helpful script just grabs the concentrations:
- Example 3.7
-
../../scripts/getboxconcs.py O3 boxEmChem19a.csv,
which results in the file ResConcs_boxEmChem19a_ O3_ppb.txt.
The do.testChems script described above is best for quickly testing and comparing different mechanisms. Usually these comparisons only involve gas-phase mechanisms such as EmChem19a or MCMv3.3Em. However, the EMEP CTM usually requires a host of extra species and reactions to accommodate secondary inorganic aerosol, sea salt, dust, organic aerosols and pollen, as discussed in Sect. 5. The EMEP CTM also requires files to specify how emissions and boundary conditions should be distributed among specific species, for example how a VOC emission should be split into C2H6, C2H4, nC4H10 etc.
In fact, GenChem produces many Fortran files which need to be copied into the appropriate ZCM_ directories, for example ZCM_EmChem19a-vbs for the scheme EmChem19a-vbs, as indicated in Table 1. The recommended way to get this directory is to use the script emep_setup.py from your temporary work directory within the box system. So, from for example box/tmp_work, do
- Example 4.1
-
./emep_setup.py EmChem19a-vbs
or just run it without arguments:
- Example 4.2
-
./emep_setup.py,
and this will provide a usage message, a debug flag and a list of the available chemical mechanisms. Users can easily edit the emep_setup.py scripts to modify the extra_mechanisms used – see Sect. S2 in the Supplement.
After emep_setup.py has successfully run, the ZCM_ directory produced contains all the files needed to run the EMEP CTM. The CM_ and CMX_ files can be copied directly to the CTM's source directory, and the EMEP CTM compiled as normal (“make clean”, “make”). The emissplit_run files need to be sent to a location specified by the user (via the EMEP CTM's emep_config.nml namelist).
We provide a number of chemical mechanisms which have been formatted for GenChem usage. These mechanisms are organised into two types, with separate directory trees:
-
base_mechanisms
These schemes are typically fairly complete sets of gas-phase photochemical mechanisms and are designed to be the core for any boxChem, ESX or EMEP CTM runs. Apart from the EMEP-developed EmChem19a, the other base schemes have been adapted from other sources for EMEP CTM usage, hence the “Em” postfix. Details of these schemes and adaptations can be found in Bergström et al. (2021a). The schemes provided with GenChem currently comprise the following (see also Table 3):
- –
EmChem19a
EmChem19a is the base EMEP chemical scheme, which has 158 gas-phase reactions in the core mechanism, and in addition to these a number of heterogeneous reactions are also included, bringing the total to 171 reactions for simple boxChem usage (cf. Table 3). This scheme is a surrogate-species scheme that has evolved over many years (Eliassen et al., 1982; Simpson et al., 1993, 2012; Bergström et al., 2021a) and has over the years been shown to compare well against other and more extensive chemical mechanisms (Kuhn et al., 1998; Andersson-Sköld and Simpson, 1999; Bergström et al., 2021a). The most recent changes have included a revised isoprene chemistry based on the CheT2 mechanism of Squire et al. (2015) and the addition of toluene and benzene as well as o-xylene to represent aromatics. A new feature of EmChem19a compared to earlier EMEP schemes is the addition of an RO2POOL species, representing the total concentration of all peroxy radicals; RO2POOL is used for setting the rates of peroxy + peroxy radical reactions. A set of new nitrate radical reactions has also been added, and reaction rates have been revised to be in line with recent International Union of Pure and Applied Chemistry (IUPAC) recommendations. For details see Bergström et al. (2021a).
- –
MCMv3.3Em
MCMv3.3Em is based on the “Master Chemical Mechanism” v3.3.1 (http://mcm.leeds.ac.uk/MCM/, last access: 6 December 2020), with a few updated reactions (mainly updates of some reaction rates to be in agreement with IUPAC recommendations from 2009 to 2018). In our implementation the MCM mechanism has over 5800 species and over 17 000 reactions. See Jenkin et al. (2015), and references therein, for details about MCM and Bergström et al. (2021a) for details about the revisions made for MCMv3.3Em. The MCM mechanism is too large for the EMEP CTM but can be run with boxChem or ESX, and it serves as an important reference mechanism.
- –
CRIv2R5Em
CRIv2R5Em is an adaption of the “Common Representative Intermediates” (CRI) scheme, with a variant of the CRI v2.2 isoprene chemistry (Jenkin et al., 2008, 2019). In order to make the scheme manageable for 3-D modelling, the full CRI scheme is reduced by only including emissions from a limited set of different VOCs (the so-called CRI_R5 reduction subset from Watson et al., 2008, is used in the EMEP adaption of CRI). Even with this reduction the CRI scheme is substantially larger than our EmChem schemes, but still well suited to 3-D modelling (see e.g. McFiggans et al., 2019, and Jenkin et al., 2019, for studies employing the CRI-R5 mechanism with the EMEP CTM). The EMEP version of CRI v2-R5 (CRIv2R5Em) is described in detail by Bergström et al. (2021a), and the revision of the isoprene chemistry by Jenkin et al. (2019).
- –
CB6r2Em
The “carbon-bond” (CB) schemes have been developed over many years as an innovative solution for dealing with chemistry in 3-D CTMs (Gery et al., 1989; Yarwood et al., 2010a, b; Luecken et al., 2019). The CB6r2 chemical scheme has been implemented without any significant change in the GenChem, except that photolysis rates have been adjusted to use MCM (for boxChem usage) or EMEP CTM surrogates. Also, the biogenic VOCs of CB6r2, ISOP (isoprene) and TERP (representing all monoterpenes, MTs), have been renamed C5H8 and APINENE (also a surrogate for all MTs in this case), respectively, since this allows the same emission reaction equation to be used for all four mechanisms if desired.
- –
-
extra_mechanisms
In this directory we store sets of reactions and sometimes species that can be appended to the base mechanisms. Many of these are essential for 3-D chemical transport modelling, whilst others are used for box model simulations. With this release we provide mechanisms for sea salt, dust, emissions from ships (EMEP uses a special ShipNOx species; see Simpson et al., 2015), and several organic aerosol and biogenic VOC (BVOC) emission options. Comments on each scheme can be found in the appropriate README files. The organic aerosol schemes will be further discussed and compared in Bergström et al. (2021b).
Tables S1–S2 in the Supplement provide brief explanations of the many currently implemented extra_mechanism packages, but we can give three important extra mechanisms as examples:
- –
BoxAero
BoxAero provides SO2 gas-phase chemistry and some reactions for very simple gas to aerosol conversion for SO3, HNO3 and N2O5. The reactions provide simplified chemical loss mechanisms for these species in the box model – they are calculated in a more complex way in the full EMEP CTM, which also includes NH3 chemistry. This directory is intended only for boxChem usage, and is applied automatically when using the do.testChems script (see Sect. 3).
- –
PM_WoodFFuelInert
PM emissions (fine and coarse) in the EMEP CTM are typically split into EC (elemental carbon), POM (primary particulate organic matter) and remaining PPM (remPPM) components. Different levels of detail are allowed, but this package enables the most common setup. POM and EC emissions are divided into those from biomass combustion and fossil fuel. POM are assumed inert, consistent with the PM_VBS_EmChem19 scheme discussed below. EC emissions are further divided into “new” and “age” components, to reflect the level of hydrophobicity (Tsyro et al., 2007; Genberg et al., 2013). In some inventories primary sulfate is also provided, represented here as pSO4 (if pSO4 is used, remPPM will then represent all PM components except EC, POM and pSO4).
- –
PM_VBS_EmChem19
PM_VBS_EmChem19 provides additional organic aerosol reactions for EmChem19a. These reactions are currently (in version rv4.36) default in the EMEP CTM and represent minor updates of the volatility-basis-set (VBS) schemes presented in Bergström et al. (2012, 2014) and Simpson et al. (2012). The default scheme used in PM_VBS_EmChem19 uses the “NPAS” version of the EMEP VBS mechanisms (with non-partitioning (i.e. inert) primary emissions and ageing of secondary organic aerosol (SOA) compounds) – see Simpson et al. (2012) (and its SI) for further details. Unlike the simple gas-phase compounds used elsewhere, SOA species are tracked as a true aerosol – with one compound representing the sum of gas- and particle-phase compounds. These semivolatile species and reaction formats are discussed more in Sects. 6.1.1 and 6.3.
- –
Table 3 summarises the number of species and reactions involved in typical boxChem or EMEP CTM usage, and Tables 4–5 give examples of the combinations of base_mechanisms and extra_mechanisms packages. When a script such as do.testChems or emep_setup.py is run (Sects. 3, 4), these scripts collect or concatenate inputs from the base directory together with those from the extra directories, to produce the input files (see Table 1) needed by GenChem. For example, running “emep_setup.py EmChem19a” would concatenate the file EmChem19a_Species.csv from the base directory with equivalent species files from PM_VBS_EmChem19, BVOC_IsoMT1_emis etc (cf. Table 5) to produce the input file GenIn_Species.txt needed by GenChem (Table 5).
Table 3Comparison of chemical mechanisms provided with GenChem 1.0, in either boxChem (mainly gas phase) or EMEP CTM (with many particle and semivolatile compounds and tracers) configurations: number of species (Ns), number of rate coefficients, (Nr, including photolysis), number of photolysis reactions (Nj), number of anthropogenic emission terms (Ne) and computer execution time (Te) for example run.
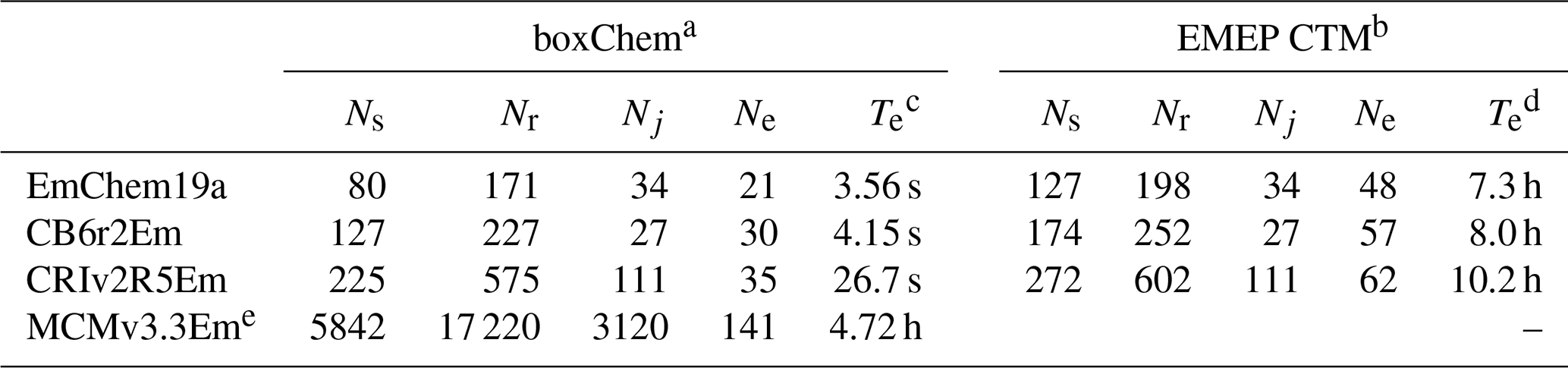
a EmChem19a-box, CB6r2Em-box and CRIv2R5Em-box; cf. Table 4. b EmChem19a-vbs, CB6r2Em-vbs and CRIv2R5Em-vbs; cf. Table 5. c boxChem execution time is for default configuration, for a 24 h run, on an x86 PC running Ubuntu-based Linux, GFortran compiler. Times given are best of five simulations. d EMEP CTM execution time is for an annual global run with regular 0.5∘ lat–long resolution grid, 20 model layers, and using 512 processors on a Linux cluster, Intel compiler. No time shown for MCM since this mechanism is too large for EMEP CTM runs. e Unlike the other provided schemes, MCMv3.3Em includes many halogen reactions. These are included for future developments. Further, MCM treats all individual reactions paths as separate reactions, whereas the other schemes frequently combine reactions into a single net reaction.
Table 4Examples of base and extra mechanisms associated with boxChem configurations.
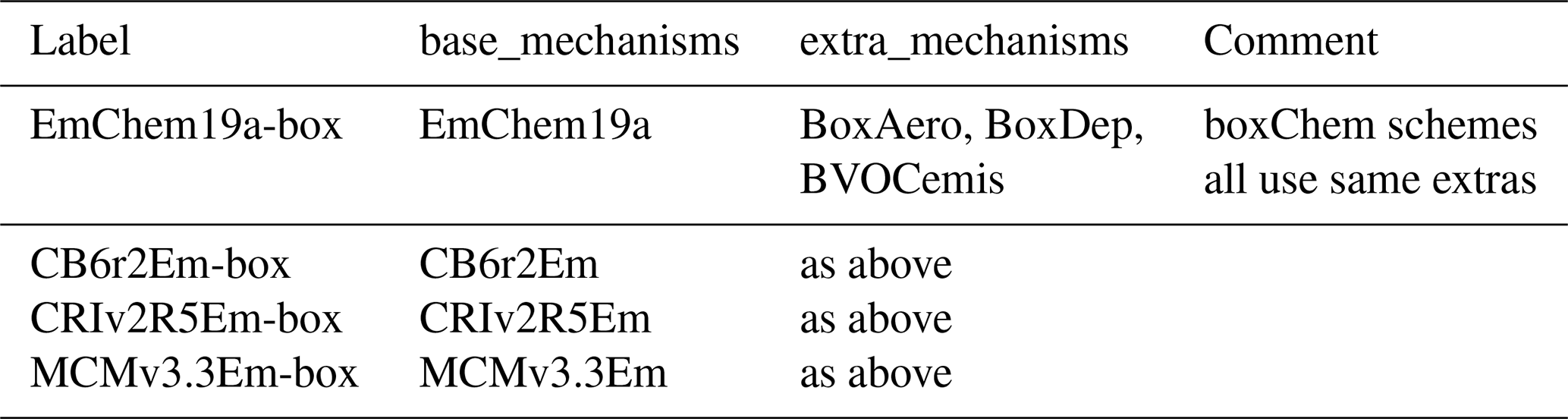
It can be seen that the GenChem system allows a very flexible approach to exploring different levels of chemical complexity, especially for EMEP CTM applications. Both base and extra mechanisms will be expanded in future GenChem versions, for example with further organic aerosol modules.
McFiggans et al. (2019)Table 5Examples of base and extra mechanisms associated with EMEP CTM configurations (via emep_setup.py).

a The simpler terms EmChem19a and EmChem19p are used in EMEP CTM rv4.35 (current at time of writing). b EMEP's default “NPAS” scheme – see Sect. 5 and Supplement Table S2. c COMMON: Aqueous, Aero2017nx, ShipNOx, PM_FFireInert, SeaSalt, DustExtended, Ash, PM_WoodFFuelInert and BVOC_SQT_NV. See Supplement Table S1 for further explanation of these packages. d Loosely based upon Hodzic et al. (2016).
Chemical mechanisms are defined in GenChem using three input files, which are themselves constructed from one or more files originating in the various sub-directories of the chem directory: GenIn_Species.csv, GenIn_Shorthands.txt and GenIn_Reactions.txt. In addition, a mechanism-specific emissplit file is needed in order to tell models how VOC emissions are to be split into individual compounds. These files are discussed below in Sects. 6.1–6.4.
6.1 Chemical species and properties: GenIn_Species.csv
The GenIn_Species.csv file is typically created by either do.testChems or emep_setup.py. As explained in Sect. 5 these scripts concatenate the Species.csv file from the base_mechanism with any “Species” files found in the specified extra_mechanisms files. The resulting GenIn_Species.csv file is a spreadsheet-friendly comma-separated file where the characteristics of the chemical compounds are given. Table 6 gives some example entries, which we briefly discuss here. Referring to Table 6, the “Spec” column is straightforward and gives the species name used in the model. The “Type” column is set to 0 for short-lived (non-advected) species, 1 for simple advected species, 2 for semivolatile compounds (see Sect. 6.1.1) and 3 for compounds that react very slowly (e.g. CH4). The chemical formula (“Formula”) is mainly for information, though it can be used to estimate the molecular weight (“MW”) if wanted, and it can be used to keep track of for example the number of carbon atoms. The MW is also not essential for many compounds, but it is needed if compounds are emitted or if outputs in mass units (e.g. µg(N) m−3) are wanted.
Table 6Example content lines from GenIn_Species.csv input file.
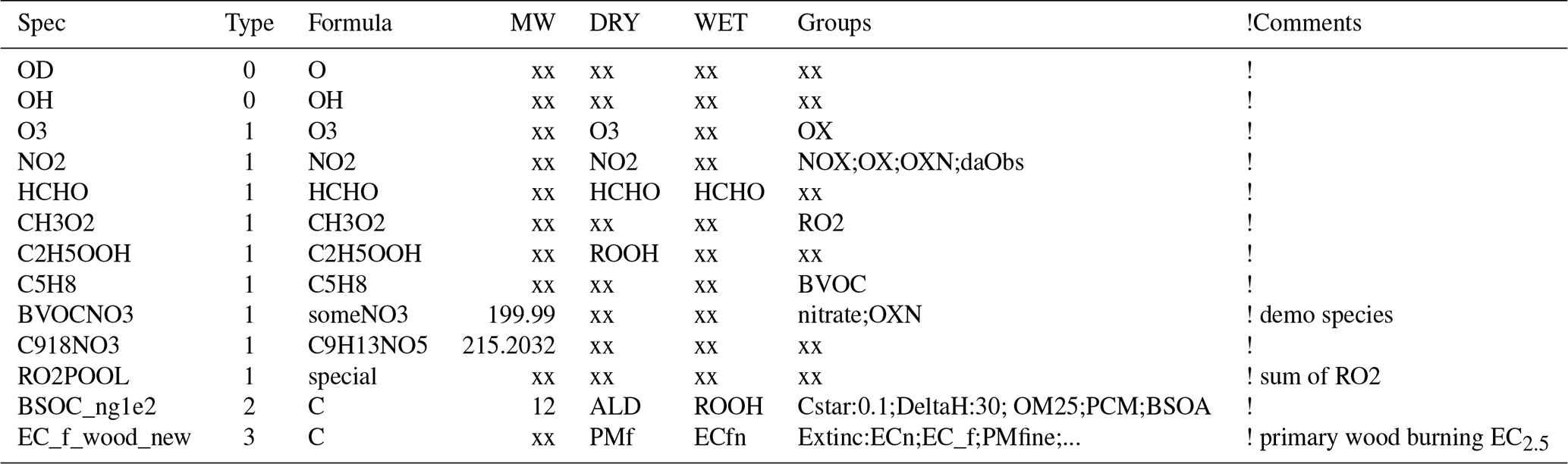
Note that commas and comment lines from file are omitted for clarity. Text after exclamation marks are for comments only and ignored by GenChem. “xx” represents dummy values.
The “DRY” and “WET” deposition columns specify which compounds undergo such deposition and which surrogate compounds are used, since the EMEP CTM calculates dry and wet deposition explicitly for only a limited number of compounds. For example, for dry deposition of O3 in the DRY column we simply use O3 since this is one of the explicitly calculated compounds, but for C2H5OOH we use the ROOH surrogate. Note that for the semivolatile SOA species the EMEP CTM will use this rate for the gas-phase fraction of the SOA; deposition of the particle phase is treated using the EMEP standard parameters for fine particles.
The “Groups” column specifies which groups the species belongs to (e.g. OXN for oxidised nitrogen and RO2 for peroxy radicals) and allows surrogate species or factors to be assigned to these groups, for example Cstar:10.0;Extinc:0.4. It is important that these groups are separated by semicolons, not commas. This rather powerful feature is discussed further in Sect. 6.1.2.
More detailed information on these entry types can be found on the readthedocs website (see Code Availability section at end).
6.1.1 Defining semivolatile species in GenIn_Species.csv
As noted above, Type 2 in GenIn_Species.csv signifies semivolatile compounds, for example SOA species. These are also subject to advection, but in addition they are semivolatile and exist in both gas and particulate phase. The EMEP CTM tracks such species by compound rather than phase and calculates the partitioning between the phases dynamically, based upon the compound's volatility (Bergström et al., 2012; Simpson et al., 2012). The approach used, the volatility basis set (VBS), follows methods developed by Donahue et al. (2006), Robinson et al. (2007) and colleagues – see Bergström et al. (2012) and references therein for details. For GenChem purposes, species labelled as Type 2 are accounted for within the list of advected species, but the start and end of the semivolatile list is calculated by GenChem.py, to produce integer variables in the Fortran code which demarcate these semivolatile compounds, for example FIRST_SEMIVOL=136 and LAST_SEMIVOL=176. (Note that GenChem will reorder different types of species to be consecutive, so despite the order of species in the GenIn_Species.csv file, all Type 2 species will lie together in the index range FIRST_SEMIVOL=136:LAST_SEMIVOL=176.)
Type 2 (SOA) species require specification of their effective saturation concentrations (C*) and the enthalpies of vaporisation (Δ Hvap), following normal VBS principles. These specifications are made using the Groups methods described next.
6.1.2 Defining Groups in GenIn_Species.csv
The Groups mechanism plays an important role for feeding key information to the EMEP CTM or boxChem. Some groups are indeed essential – for example, in EmChem19a, CRIv2R5Em and MCMv3.3Em, the RO2_GROUP needs to be set correctly to get the correct concentrations of the special RO2POOL concentration, and the deposition of groups of compounds (e.g. oxidised nitrogen deposition) depends on those compounds being correctly identified by their groups. Groups have to be separated by a semicolon, and there are two classes of group labels for a specific species:
- i.
simple name; for example OXN indicates here that this species belongs to the group of oxidised nitrogen compounds. In the CTM code, the members of one group are easily accessible so they can be treated specially (see Sect. 7.3).
- ii.
compound groups, which specify numerical or character values to pass specific properties, for example the groups Cstar:0.1 and DeltaH:30 for BSOC_ng100, or Extinc:ECn for EC_f_wood_new in Table 6.
If a species is both a member of a class (i) group and has a (wet or dry) deposition surrogate, additional WDEP or DDEP groups will be automatically generated, for example DDEP_OXN_GROUP.
The specification of numerical or character values (group class ii) is indicated with colon notation (as opposed to the semicolon used to separate groups). For example, SOA species which use the VBS system require specification of their effective saturation concentrations at 298 K (C*, in µg m−3) and the vaporisation enthalpies (Δ Hvap, in kJ mol−1), and for aerosol optical absorption we need extinction coefficients. These specifications are simply set through the groupings Cstar, DeltaH and Extinc, with for example Cstar:1.0e-2;DeltaH:30.0 for SOA and Extinc:SO4 for sulfate. Section 7.3 provides further explanation of such groups.
These groupings are not hard-coded in GenChem and may or may not be used by any CTM, so this system provides an easily extensible mechanism for introducing new characteristics into modelling systems.
6.2 Defining chemical reactions, A) GenIn_Shorthands.txt
Firstly, we can note that in the EMEP CTM and boxChem systems some key variables have special names and can be used in either the GenIn_Shorthands.txt or the GenIn_Reactions.txt files. The variables which are known to the EMEP CTM and GenChem codes are TEMP (temperature); TINV (inverse temperature); and M, O2, N2 and H2O (concentrations of air, oxygen, nitrogen and water vapour, respectively).
Other shorthand notation is often used in GenIn_Reactions.txt (Sect. 6.3), and this has to be defined first in the GenIn_Shorthands.txt file. For example, we might use KHO2RO2 as a generic rate coefficient for HO2+RO2 reactions. The shorthand LogTdiv300 is also frequently used for the common expression log(TEMP/300). The GenIn_Shorthands.txt file is typically created by either do.testChems or emep_setup.py, and these scripts concatenate the file generic_Shorthands.txt from the chem directory and the chemistry-specific files, for example chem/base_mechanisms/EmChem19a_Shorthands.txt, to produce the needed GenChem input file.
The name and the expression for a shorthand have to be separated by whitespace for GenChem to process it. Names of species can also be used in shorthand expressions. Figure 4 illustrates several examples, including how shorthand expressions defined earlier can be re-used in the same system – as done for the MCM example in producing the KMT08 variable. The last example, for KMT3, also illustrates that the right-hand side can be a function, which of course needs to be compatible with the Fortran code of the calling code.

6.3 Defining chemical reactions, B) GenIn_Reactions.txt
The GenIn_Reactions.txt file is typically created
by either do.testChems or emep_setup.py,
which concatenate the appropriate Reactions.txt file
from the wanted chem/base_mechanisms directory
(e.g. chem/base_mechanisms/EmChem19a_Reactions.txt)
and those from the wanted chem/extra_mechanisms directories.
The GenIn_Reactions.txt file contains the chemical reactions, with
the following format:
![]() and with the reaction consisting of reactants and products, along
with their stoichiometric factors as appropriate. The semicolon marks
the end of the reactions, and whitespace is needed between all terms,
for example between a stoichiometric factor and a species. Some typical lines
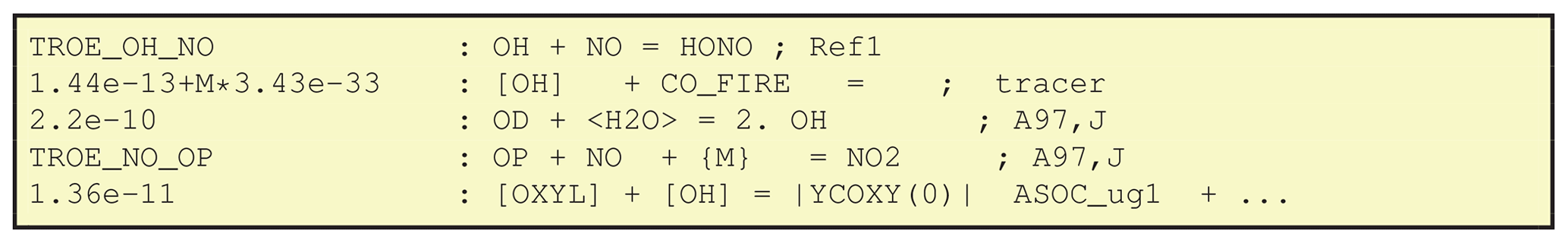
are given in Fig. 5. The first line here is trivial,
in the sense that OH, NO and HONO are all normal chemical species as
defined in GenIn_Species.csv, and GenChem will add
production and loss terms appropriately for each, with a reaction rate
given by the TROE_OH_NO shorthand.
and with the reaction consisting of reactants and products, along
with their stoichiometric factors as appropriate. The semicolon marks
the end of the reactions, and whitespace is needed between all terms,
for example between a stoichiometric factor and a species. Some typical lines
are given in Fig. 5. The first line here is trivial,
in the sense that OH, NO and HONO are all normal chemical species as
defined in GenIn_Species.csv, and GenChem will add
production and loss terms appropriately for each, with a reaction rate
given by the TROE_OH_NO shorthand.
GenChem is flexible as to whether products are written explicitly or with stoichiometric coefficients (i.e. 2 OH is the same as OH + OH). Non-integer stoichiometric coefficients are allowed, since we often condense multiple branches of a reaction into one equation for CTM use.
6.3.1 The funny brackets [], <>, {} and : constant, fixed and helper species, and yield modifiers
Four types of brackets are used in GenChem_Reactions.txt files to modify the way compounds or yields are handled:
- [ constant_species ]:
-
The second reaction of Fig. 5 illustrates a nice feature of GenChem; concentrations of species such as OH which are key parts of the photochemical reaction mechanism can be treated as constants over a time step in this particular reaction. In this way tracers are easily added. In this example the CO_FIRE tracer is emitted along with the “real” CO, but its existence is not allowed to affect the standard photochemistry. The [] around the OH signify that this CO tracer has a chemical loss due to OH, but that we do not allow the model's OH to be degraded by this artificial species. The lack of products in this example also signifies that we do not track the products of this loss, just the CO_FIRE itself.
- < fixed_species >:
-
The third example from Fig. 5 illustrates the use of <> notation. In this case, the species within the angle brackets is not one of the chemical compounds tracked or changed by the chemical solver but is rather a compound whose concentration is effectively constant during a simulation time step and set by the EMEP and/or boxChem code, based upon humidity and pressure from the meteorological model. (In the KPP system these species are defined as “DEFFIX” compounds; Sandu and Sander, 2006.) The compounds used so far in this way are H2O, N2, O2 and M (air molecules). This last line could equivalently have been written:
- Example 6.1
-
2.2e-10*H2O : OD = 2. OH ;,
where the H2O concentration now applies as a simple part of the rate coefficient. (This is actually exactly the way GenChem handles this internally.)
- { helper_species }:
-
The fourth reaction in Fig. 5 shows another type of special notation. Species within curly brackets are not used in any way, but they can be added to the reactions as helpful comments illustrating reactants whose concentrations are already included in the rate expression – in the example the TROE_NO_OP rate expression takes into account the pressure (i.e. [M]) dependence for this three-body reaction.
- | yield-modifier |:
-
The final reaction of Fig. 5 illustrates the use of brackets. These are sometimes used in the reaction schemes for secondary organic aerosol, as seen here for the production of the semivolatile ASOC_ug1 species. The contents of represent yield expressions which will be updated each time step in the chemical solver. The output CM_Reactions2.inc file for this case includes the term:
- Example 6.2
-
P = YCOXY(0)* 1.36e-11 * xnew(OXYL) * xnew(OH).
These variables (here YCOXY(0)) must be predefined in the appropriate Fortran system (boxChem or EMEP CTM) in order for
box_setup.pyoremep_setup.pyto achieve successful compilation.
6.3.2 Adding emissions in GenIn_Reactions.txt
When using emissions in GenIn_Reactions.txt, the labels used for associated emission files have to be defined in a special line, for example “emisfiles:sox,nox,co,voc,nh3” as in the first line of Fig. 6. These emission labels (e.g. lower-case nox) are those used in EMEP for emission input files, as well as being the file endings for the respective emissplit file (see Sect. 6.4). Other labels can easily be used and defined as long as the emissplit system exists to convert these groups into model species (e.g. nox into NO and NO2, or voc into C2H6, NC4H10 etc.).

The next three lines in Fig. 6 are examples for emission reactions: the reaction rate is denoted as rcemis(SPECIES,KDIM), and there is no reactant in the reaction. GenChem will replace KDIM with the vertical coordinate, assumed to be k, in the Fortran code, e.g. giving rcemis(NO,k).
BVOC emissions are special, in that specific functions exist in the EMEP model for dealing with the light, temperature and other dependencies of these (see e.g. Simpson et al., 2012). In the extra_mechanisms files used for the EMEP CTM setup, we use the “rcbio” functions as shown in Fig. 7.

Figure 7Biogenic emissions lines from input files used in GenIn_Reactions.txt, for either EMEP or boxChem setups.
When using boxChem, a very simplified system is used for BVOC emissions, illustrated also in Fig. 7. The SUN variable (borrowed from the KPP system) allows for simple variation of emissions with zenith angle (and gives zero emissions at night, when SUN = 0, and maximal emissions at noon, when SUN = 1). The numerical coefficients (5.0e7 or 2.5e6) correspond to typical emission rates (units are molecules cm3 s−1; see the appropriate mechanism file for species), and the fIsop, fMTL and fMTP factors, which are set in config_box.nml, provide the possibility to scale emissions of isoprene, light-dependent monoterpenes and light-independent (“pool”) monoterpenes, respectively. For example, light-dependent emissions for MT can be reduced by 50 % using fMTL = 0.5, and pool MT emissions can be set to 0 by setting fMTP = 0.0.
6.3.3 Adding photolysis reactions in GenIn_Reactions.txt
The reaction rates of photolysis reactions are denoted as rcphot(PHOT_ID). GenChem will automatically add the “k” dependency on the vertical level to the photolysis rate (e.g. to give rcphot(IDH2O2,k)). The index variables (e.g. IDO3_O1D) refer to photolysis rates as defined in the EMEP and boxChem codes.
6.4 Emissions speciation: emissplit files
Emissions are often provided to models for groups of compounds, for example NOx for NO and NO2, and NMVOC for non-methane volatile organic compounds. These emissions need to be assigned to individual chemical compounds and converted from mass to number using the appropriate molecular weight.
The default files for SOx, NOx, CO and NH3 are identical across all provided schemes and provided in the input/emissplit_defaults directory as files (in lower case) such as emissplit_defaults_nox.csv. For NMVOC and PM inventories specific files are needed for each chemical mechanism, sometimes depending on available inventories.
Default NMVOC emission splits are provided in GenChem for 11 different source categories (covering traffic, agriculture etc.), according to the so-called “SNAP” classifications (Selected Nomenclature for sources of Air Pollution, EEA, 2007, as used in the EMEP model, Simpson et al., 2012). The provided data are based upon average UK emission profiles from Passant (2002) and emissions from 2010, and they have been adapted in this work for each base chemistry scheme.
For GenChem we provide such NMVOC files for all supported chemical mechanisms, in the appropriate directory. Thus, for EmChem19a the file for NMVOC emissions would be named EmChem19a_emissplit_defaults_voc.csv. For boxChem testing the do.GenChem or do.testChems script will move this file to inputs/emissplit_run and also copy it to ZCMBOX_EmChem19a/emissplit_run and rename it to simply emissplit_defaults_voc.csv. If emep_setup.py is used, the emissplit_run directory is copied to ZCM_EmChem19a/emissplit_run.
In the EMEP CTM it is common for these default values to be overridden by “emissplit_specials” files which can assign country- and sector-specific NOx, NMVOC and PM profiles. Such profiles need to be generated by the EMEP CTM user, however, and are not part of GenChem. An example of this system, and such emission splits, is given in the Supplement, Table S5, of Simpson et al. (2012). Also in boxChem, users may of course modify any of these emissplit files – see Sect. S2.3 in the Supplement.
The main purpose of the GenChem system is to convert chemical equations into Fortran code suitable for use in the boxChem and/or EMEP CTM systems. The output files, prefixed with CM_ to denote chemical mechanism, are summarised in Table 1 and discussed in the relevant section below.
7.1 CM_ChemDims_mod.f90 – the dimensions of the chemical system
The CM_ChemDims_mod module provides basic information (Fig. 8) about the dimensioning of the chemical system, giving for example the total number of species (NSPEC_TOT), photolysis rates (NPHOTOLRATES) or emission files (NEMIS_File).

Figure 8Selected lines from the output file CM_ChemDims_mod.f90. (The actual file has comments for each entry.)
7.2 CM_ChemSpecs_mod.f90 – chemical compound information
This file specifies basic information about the chemical compounds, in terms of number, indices and some chemical characteristics. Extracts of the file are shown in Figs. 9 and 10. As seen in Fig. 9, this module provides the simple indices which represent the chemical compounds in the EMEP systems, for example OH = 3. Indices are additionally provided for the short-lived and advected species (IXSHL_ indices and IXADV_ indices).

One subroutine, define_chemicals, is generated in this module, which sets the contents of a Fortran-derived type array named “species”. The derived species type (cf. Fig. 10) contains the following elements: name, molwt, nmhc, carbons, nitrogen and sulphurs. This routine is called in the initialisation of the CTM, and thereafter this array provides useful information on each species; for example species(HNO3)%molwt is the molecular weight of HNO3, and species(C5H8)%carbons the number of carbon atoms in C5H8. The nmhc element identifies if a species is a non-methane hydrocarbon (nmhc = 1) or not (nmhc = 0). This file also defines pointers to the advected, short-lived and semivolatile species list, which is used in the EMEP CTM.

Figure 10Selected lines from the species array defined in the GenChem-produced file CM_ChemSpecs_mod.f90.
7.3 CM_ChemGroups_mod.f90 – Fortran arrays for the groups
As noted in Sect. 6.1.2, GenChem makes use of the information provided in the Groups column of GenIn_Species.txt to produce Fortran arrays; for example OXN_GROUP would include all oxidised nitrogen compounds specified with OXN in the Groups column (see Fig. 11 for more examples). These groups are also accessible through a Fortran pointer system, for example
chemgroups(1)%name="RO2"chemgroups(1)%ptr=>RO2_GROUP...chemgroups(7)%name="DDEP_OXN"chemgroups(7)%ptr=>DDEP_OXN_GROUP
which allows the EMEP CTM to access and perform actions on these groups without having to “know” the names of the species involved. For example, dry deposition of all OXN species can be formulated in the model either as a simple loop over all the compounds in DDEP_OXN_GROUP_OXN or, equivalently, by finding the index of DDEP_OXN in chemgroups and following the Fortran pointer to the array of indices.

The more complex class (ii) compound groups discussed in Sect. 6.1.2, in which numbers or character strings are associated with a group, result in further arrays in CM_ChemGroup_mod.f90 which provide access to these data. Fig. 12 illustrates some examples of this.

7.4 CM_DryDep.inc and CM_WetDep.inc – mapping individual chemical species to dry- and wet-deposition surrogates
Species which have had dry- or wet-deposition surrogates specified in the DRY or WET column of GenIn_Species.txt are listed as part of a Fortran-derived type (depmap) in the output files CM_DryDep.inc and CM_WetDep.inc using code such as in Fig. 13, where the first entry is the index of the species in the list of advected species and the second entry is the surrogate species as discussed in Sect. 6.1. The last entry can be used to set fixed values of deposition velocity (although this is not typically done for the EMEP CTM). This listing is then used by the EMEP CTM as part of the standard deposition calculations.

7.5 CM_Reactions1(2).inc and CM_Reactions.log – the chemical reactions code
Section 1 and Fig. 1 have already presented an example section from the file CM_Reactions1.inc. This file generally comprises such production and loss terms (P and L, respectively) for all, or the majority, of the model's chemically reacting species. The terms xold and xnew in this example represent the original and updated concentrations arising from the TWOSTEP Gauss–Seidel calculations for the time step dt2. For slowly reacting species such as CH4, a second file (CM_Reactions2.inc) is produced with the same type of equations, but which can lie outside of the iteration loop in the EMEP system; for example the EMEP CTM code can be summarised as

The file CM_Reactions.log is not needed by the EMEP CTM, but it is output as a valuable help file, containing a listing of all the chemical reactions, along with their index in the “rct”, “rcphot” and “rcemis” arrays as appropriate. Equations which were specified with simple constant values (e.g. 1.0e-5) are also reported here with the k indicator and rate.
7.6 CM_ChemRates_mod.f90 – setting the reaction rates
CM_ChemRates_mod.f90 is the module where all chemical rate coefficients and photolysis rates are calculated (this is done in every advection step in the EMEP CTM for example). The module is entirely written by GenChem and produces two subroutines:
-
setChemRates
-
setPhotolUsed.
Typical lines of CM_ChemRates_mod.f90 are shown in Fig. 14. This figure also illustrates how the model makes use of the defined meteorology-associated arrays TEMP, TINV, RH and LOG300DIVT (Sect. 6.2). The setPhotolUsed array routine is much simpler, in that it just lists the indices used, for example
- Example 7.1
-
photol_used = (/ IDO3_O3,IDO3_O1D, ... /).

7.7 CM_EmisFile.inc – listing emission files
The last file simply lists the names used for input emission files. By tradition the EMEP system has used lower case for these emission markers (cf. Fig. 15). The names used are triggered by the “emisfiles:” line of GenIn_Reactions.txt as discussed in Sect. 6.3. Other typical emissions that might be used, depending on the application and available inventories, are pm25 (fine particulate matter), ec (elemental carbon), oc (organic carbon) or pom (primary organic matter).

This paper outlines the structure and usage of the GenChem system, which includes a chemical pre-processor (GenChem.py) for converting chemical equations into differential form for use in atmospheric chemical transport models (CTMs). Although primarily intended for users of the EMEP CTM and related systems, GenChem also features a simple box-model testing system (boxChem), which can run as a stand-alone chemical solver, enabling for example easy testing of chemical mechanisms against each other. GenChem has been developed and tested in a Linux environment, but it can be run in virtual environments on Windows or other architectures.
The mechanisms included now reflect those used or made available for the EMEP CTM, as well as the MCM scheme which works in the boxChem mode. The EmChem19a-vbs scheme is the default mechanism used in the EMEP CTM, but we include slightly adapted versions of CB6, which is used in the widely used CAMx model (http://www.camx.com, last access: 2 December 2020) and CMAQ (Luecken et al., 2019), and the CRIv2-R5 scheme, which is used in STOCHEM (Archibald et al., 2010; Khan et al., 2015). It is hoped that some of the other widely used mechanisms can be added in future, for example the MOZART scheme (Emmons et al., 2010; Surendran et al., 2015); the RACM scheme (Stockwell et al., 1997; Goliff et al., 2013); or SAPRC-07 (Carter, 2010), which is also used in CMAQ (https://www.airqualitymodeling.org/index.php/CMAQv5.1_Mechanisms, last access: 2 December 2020).
As provided here, GenChem is already a useful tool for exploring different chemical mechanisms, both for gas-phase and simple (EMEP-compatible) aerosol-phase systems. For example, Fig. 3 showed one comparison between the EMEP CTM's EmChem19a scheme and the far-more-advanced CRIv2R5Em and MCMv3.3Em schemes, and Bergström et al. (2021a) provide many more. Such comparisons will greatly aid the development of new EMEP mechanisms, or indeed comparison of any mechanisms of interest to users.
In future new and/or updated chemical mechanisms will be added as well as utility scripts to simplify result analysis, and to convert between GenChem and other formats, such as those used in KPP (Damian et al., 2002), the MCM website, FACSIMILE (https://www.mcpa-software.com/, last access: 2 December 2020; http://mcm.york.ac.uk/tutorial_sec1.htt, last access: 2 December 2020) or more recently PyBox (Topping et al., 2018).
The code needed to run the GenChem system is released as open-source code under the GNU license (https://github.com/metno/genchem, last access: 2 December 2020), and GenChem v1.0 as documented here is archived on Zenodo (https://doi.org/10.5281/zenodo.4133566, Simpson et al., 2020b). The GenChem user guide is provided at https://genchem.readthedocs.io (last access: 2 December 2020). The EMEP MSC-W CTM can be found at https://doi.org/10.5281/zenodo.4230110 (EMEP MSC-W, 2020).
The supplement related to this article is available online at: https://doi.org/10.5194/gmd-13-6447-2020-supplement.
DS wrote the original Perl version of the GenChem script and has written much of the Fortran and Python code of the more extensive GenChem and boxChem systems. AB wrote the first Python version of GenChem.py. RB worked with the chemical mechanisms. HI and JJ worked with and improved many scripts from the GenChem system, including conversion of CRI and MCM codes to GenChem format. MP added the Docker functionality and helped test the code on Windows. AV added the Pollen module and also contributed to some code developments. All authors contributed to the paper.
The authors declare that they have no conflict of interest.
We thank Garry D. Hayman, Centre for Ecology & Hydrology, UK; Michael E. Jenkin, Atmospheric Chemistry Services, UK; Greg Yarwood, Ramboll, USA; and Andrew Rickard and colleagues from the MCM website (http://mcm.york.ac.uk/, last access: 2 December 2020) for making available the chemical mechanism codes and data which underlie the GenChem system.
This research has been supported by the European Commission's Seventh Framework Programme (FP7; ECLAIRE (grant no. 282910)); the EMEP under UN-ECE; EMEP under UN-ECE; the Swedish Strategic Research project MERGE; the FORMAS BVOC-SIE project; the Swedish Research Council project Photosmog (639-2013-6917); and the UK Dept. for Environ., Food and Rural Affairs.
This paper was edited by Christoph Knote and reviewed by two anonymous referees.
Andersson-Sköld, Y. and Simpson, D.: Comparison of the chemical schemes of the EMEP MSC-W and the IVL photochemical trajectory models, Atmos. Environ., 33, 1111–1129, 1999. a
Archibald, A. T., Cooke, M. C., Utembe, S. R., Shallcross, D. E., Derwent, R. G., and Jenkin, M. E.: Impacts of mechanistic changes on HOx formation and recycling in the oxidation of isoprene, Atmos. Chem. Phys., 10, 8097–8118, https://doi.org/10.5194/acp-10-8097-2010, 2010. a
Ashworth, K., Chung, S. H., Griffin, R. J., Chen, J., Forkel, R., Bryan, A. M., and Steiner, A. L.: FORest Canopy Atmosphere Transfer (FORCAsT) 1.0: a 1-D model of biosphere–atmosphere chemical exchange, Geosci. Model Dev., 8, 3765–3784, https://doi.org/10.5194/gmd-8-3765-2015, 2015. a, b
Bergström, R., Denier van der Gon, H. A. C., Prévôt, A. S. H., Yttri, K. E., and Simpson, D.: Modelling of organic aerosols over Europe (2002–2007) using a volatility basis set (VBS) framework: application of different assumptions regarding the formation of secondary organic aerosol, Atmos. Chem. Phys., 12, 8499–8527, https://doi.org/10.5194/acp-12-8499-2012, 2012. a, b, c
Bergström, R., Hallquist, M., Simpson, D., Wildt, J., and Mentel, T. F.: Biotic stress: a significant contributor to organic aerosol in Europe?, Atmos. Chem. Phys., 14, 13643–13660, https://doi.org/10.5194/acp-14-13643-2014, 2014. a
Bergström, R., Jenkin, M., Hayman, G., and Simpson, D.: Update and comparison of atmospheric chemistry mechanisms for the EMEP MSC-W model system – EmChem19a, EmChem19X, CRIv2R5Em, CB6r2Em, and MCMv3.3Em, in preparation, 2021a. a, b, c, d, e, f, g
Bergström, R., et al.: Organic aerosol schemes for the EMEP MSC-W model for European and Global scale simulations, in preparation, 2021b. a
Carter, W. P. L.: Development of the SAPRC-07 chemical mechanism, Atmos. Environ., 44, 5324–5335, https://doi.org/10.1016/j.atmosenv.2010.01.026, 2010. a
Damian, V., Sandu, A., Damian, M., Potra, F., and Carmichael, G.: The kinetic preprocessor KPP – a software environment for solving chemical kinetics, Computers Chem Eng., 26, 1567–1579, https://doi.org/10.1016/S0098-1354(02)00128-X, 2002. a, b
Donahue, N. M., Robinson, A. L, Stanier, C. O., and Pandis, S. N.: Coupled Partitioning, Dilution, and Chemical Aging of Semivolatile Organics, Environ. Sci. Technol., 40, 2635–2643, https://doi.org/10.1021/es052297c, 2006. a
EEA: Atmospheric Emissions Inventory Guidebook, 2nd edn., EEA (European Environment Agency), Copenhagen, Denmark, Technical report No. 16/2007, ISBN 978-92-9167-972-0, available at: https://www.eea.europa.eu/publications/EMEPCORINAIR5 (last access: 2 December 2020), 2007. a
Eliassen, A., Hov, Ø., Isaksen, I., Saltbones, J., and Stordal, F.: A Lagrangian long-range transport model with atmospheric boundary layer chemistry, J. Appl. Met., 21, 1645–1661, 1982. a
Eller, P., Singh, K., Sandu, A., Bowman, K., Henze, D. K., and Lee, M.: Implementation and evaluation of an array of chemical solvers in the Global Chemical Transport Model GEOS-Chem, Geosci. Model Dev., 2, 89–96, https://doi.org/10.5194/gmd-2-89-2009, 2009. a
EMEP MSC-W: Open Source EMEP/MSC-W model rv4.36 (202011), Zenodo, https://doi.org/10.5281/zenodo.4230110, 2020. a
Emmons, L. K., Walters, S., Hess, P. G., Lamarque, J.-F., Pfister, G. G., Fillmore, D., Granier, C., Guenther, A., Kinnison, D., Laepple, T., Orlando, J., Tie, X., Tyndall, G., Wiedinmyer, C., Baughcum, S. L., and Kloster, S.: Description and evaluation of the Model for Ozone and Related chemical Tracers, version 4 (MOZART-4), Geosci. Model Dev., 3, 43–67, https://doi.org/10.5194/gmd-3-43-2010, 2010. a
Genberg, J., Denier van der Gon, H. A. C., Simpson, D., Swietlicki, E., Areskoug, H., Beddows, D., Ceburnis, D., Fiebig, M., Hansson, H. C., Harrison, R. M., Jennings, S. G., Saarikoski, S., Spindler, G., Visschedijk, A. J. H., Wiedensohler, A., Yttri, K. E., and Bergström, R.: Light-absorbing carbon in Europe – measurement and modelling, with a focus on residential wood combustion emissions, Atmos. Chem. Phys., 13, 8719–8738, https://doi.org/10.5194/acp-13-8719-2013, 2013. a
Gery, M., Whitten, G., Killus, J., and Dodge, M.: A photochemical kinetics mechanism for urban and regional scale computer modelling, J. Geophys. Res., 94, 12925–12956, 1989. a
Goliff, W. S., Stockwell, W. R., and Lawson, C. V.: The regional atmospheric chemistry mechanism, version 2, Atmos. Environ., 68, 174–185, https://doi.org/10.1016/j.atmosenv.2012.11.038, 2013. a
Hertel, O., Christensen, J., Runge, E., Asman, W., Berkowicz, R., Hovmand, M., and Hov, O.: Development and Testing of a New Variable Scale Air-Pollution Model – ACDEP, Atmos. Environ., 29, 1267–1290, 1995. a
Hodzic, A., Kasibhatla, P. S., Jo, D. S., Cappa, C. D., Jimenez, J. L., Madronich, S., and Park, R. J.: Rethinking the global secondary organic aerosol (SOA) budget: stronger production, faster removal, shorter lifetime, Atmos. Chem. Phys., 16, 7917–7941, https://doi.org/10.5194/acp-16-7917-2016, 2016. a
Jenkin, M. E., Watson, L. A., Utembe, S. R., and Shallcross, D. E.: A Common Representative Intermediates (CRI) mechanism for VOC degradation. Part 1: Gas phase mechanism development, Atmos. Environ., 42, 7185–7195, https://doi.org/10.1016/j.atmosenv.2008.07.028, 2008. a
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015. a
Jenkin, M. E., Khan, M. A. H., Shallcross, D. E., Bergström, R., Simpson, D., Murphy, K. L. C., and Rickard, A. R.: The CRI v2.2 reduced degradation scheme for isoprene, Atmos. Environ., 212, 172–182, https://doi.org/10.1016/j.atmosenv.2019.05.055, 2019. a, b, c
Jeričević, A., Kraljević, L., Grisogono, B., Fagerli, H., and Večenaj, Ž.: Parameterization of vertical diffusion and the atmospheric boundary layer height determination in the EMEP model, Atmos. Chem. Phys., 10, 341–364, https://doi.org/10.5194/acp-10-341-2010, 2010. a
Karl, M., Castell, N., Simpson, D., Solberg, S., Starrfelt, J., Svendby, T., Walker, S.-E., and Wright, R. F.: Uncertainties in assessing the environmental impact of amine emissions from a CO2 capture plant, Atmos. Chem. Phys., 14, 8533–8557, https://doi.org/10.5194/acp-14-8533-2014, 2014. a
Khan, M. A. H., Cooke, M. C., Utembe, S. R., Archibald, A. T., Derwent, R. G., Jenkin, M. E., Morris, W. C., South, N., Hansen, J. C., Francisco, J. S., Percival, C. J., and Shallcross, D. E.: Global analysis of peroxy radicals and peroxy radical-water complexation using the STOCHEM-CRI global chemistry and transport model, Atmos. Environ., 106, 278–287, https://doi.org/10.1016/j.atmosenv.2015.02.020, 2015. a
Kuhn, M., Builtjes, P., Poppe, D., Simpson, D., Stockwell, W., Andersson-Sköld, Y., Baart, A., Das, M., Fiedler, F., Hov, Ø., Kirchner, F., Makar, P., Milford, J., Roemer, M., Ruhnke, R., Strand, A., Vogel, B., and Vogel, H.: Intercomparison of the gas-phase chemistry in several chemistry and transport models, Atmos. Environ., 32, 693–709, 1998. a
Langner, J., Bergström, R., and Pleijel, K.: European scale modeling of sulfur, oxidized nitrogen and photochemical oxidants. Model development and evaluation for the 1994 growing season,Swedish Meteorological and Hydrological Institute, Norrköping, Sweden, SMHI Reports Meteorology and Climatology RMK No. 82, 71 pp., 1998. a
Lowe, D., Topping, D., and McFiggans, G.: Modelling multi-phase halogen chemistry in the remote marine boundary layer: investigation of the influence of aerosol size resolution on predicted gas- and condensed-phase chemistry, Atmos. Chem. Phys., 9, 4559–4573, https://doi.org/10.5194/acp-9-4559-2009, 2009. a
Lowe, D., Ryder, J., Leigh, R., Dorsey, J. R., and McFiggans, G.: Modelling multi-phase halogen chemistry in the coastal marine boundary layer: investigation of the relative importance of local chemistry vs. long-range transport, Atmos. Chem. Phys., 11, 979–994, https://doi.org/10.5194/acp-11-979-2011, 2011. a
Luecken, D. J., Yarwood, G., and Hutzell, W. T.: Multipollutant modeling of ozone, reactive nitrogen and HAPs across the continental US with CMAQ-CB6, Atmos. Environ., 201, 62–72, https://doi.org/10.1016/j.atmosenv.2018.11.060, 2019. a, b
Makar, P., Fuentes, J., Wang, D., Staebler, R., and Wiebe, H.: Chemical processing of biogenic hydrocarbons within and above a temperate deciduous forest, J. Geophys. Res., 104, 3581–3603, 1999. a
McFiggans, G., Mentel, T. F., Wildt, J., Pullinen, I., Kang, S., Kleist, E., Schmitt, S., Springer, M., Tillmann, R., Wu, C., Zhao, D., Hallquist, M., Faxon, C., Le Breton, M., Hallquist, A. M., Simpson, D., Bergstrom, R., Jenkin, M. E., Ehn, M., Thornton, J. A., Alfarra, M. R., Bannan, T. J., Percival, C. J., Priestley, M., Topping, D., and Kiendler-Scharr, A.: Secondary organic aerosol reduced by mixture of atmospheric vapours, Nature, 565, 587–593, https://doi.org/10.1038/s41586-018-0871-y, 2019. a, b
Murphy, B. N., Donahue, N. M., Fountoukis, C., and Pandis, S. N.: Simulating the oxygen content of ambient organic aerosol with the 2D volatility basis set, Atmos. Chem. Phys., 11, 7859–7873, https://doi.org/10.5194/acp-11-7859-2011, 2011. a
Omstedt, A., Edman, M., Claremar, B., and Rutgersson, A.: Modelling the contributions to marine acidification from deposited SOx, NOx, and NHx in the Baltic Sea: Past and present situations, Continental Shelf Res., 111, 234–249, https://doi.org/10.1016/j.csr.2015.08.024, 2015. a
Ots, R., Heal, M. R., Young, D. E., Williams, L. R., Allan, J. D., Nemitz, E., Di Marco, C., Detournay, A., Xu, L., Ng, N. L., Coe, H., Herndon, S. C., Mackenzie, I. A., Green, D. C., Kuenen, J. J. P., Reis, S., and Vieno, M.: Modelling carbonaceous aerosol from residential solid fuel burning with different assumptions for emissions, Atmos. Chem. Phys., 18, 4497–4518, https://doi.org/10.5194/acp-18-4497-2018, 2018. a
Passant, N.: Speciation of UK emissions of non-methane volatile organic compounds,AEA Technology, Culham, Abingdon, United Kingdom, Report ENV-0545 ENV-0545, 289 pp., 2002. a
Robinson, A. L., Donahue, N. M., Shrivastava, M. K., Weitkamp, E. A., Sage, A. M., Grieshop, A. P., Lane, T. E., Pierce, J. R., and Pandis, S. N.: Rethinking Organic Aerosols: Semivolatile Emissions and Photochemical Aging, Science, 315, 1259–1262, https://doi.org/10.1126/science.1133061, 2007. a
Sandu, A. and Sander, R.: Technical note: Simulating chemical systems in Fortran90 and Matlab with the Kinetic PreProcessor KPP-2.1, Atmos. Chem. Phys., 6, 187–195, https://doi.org/10.5194/acp-6-187-2006, 2006. a, b
Sandu, A., Verwer, J. G., Blom, J. G., Spee, E. J., Carmichael, G. R., and Potra, F. A.: Benchmarking stiff ODE solvers for atmospheric chemistry problems 2. Rosenbrock solvers, Atmos. Environ., 31, 3459–3472, 1997. a, b
Simpson, D.: Biogenic emissions in Europe 2: Implications for ozone control strategies, J. Geophys. Res., 100, 22891–22906, 1995. a
Simpson, D. and Tuovinen, J.-P.: ECLAIRE Ecosystem Surface Exchange model (ESX), in: Transboundary particulate matter, photo-oxidants, acidifying and eutrophying components, Status Report 1/2014, The Norwegian Meteorological Institute, Oslo, Norway, 147–154, 2014. a
Simpson, D., Andersson-Sköld, Y., and Jenkin, M. E.: Updating the chemical scheme for the EMEP MSC-W oxidant model: current status, The Norwegian Meteorological Institute, Oslo, Norway, EMEP MSC-W Note 2/93, 33 pp., 1993. a
Simpson, D., Benedictow, A., Berge, H., Bergström, R., Emberson, L. D., Fagerli, H., Flechard, C. R., Hayman, G. D., Gauss, M., Jonson, J. E., Jenkin, M. E., Nyíri, A., Richter, C., Semeena, V. S., Tsyro, S., Tuovinen, J.-P., Valdebenito, Á., and Wind, P.: The EMEP MSC-W chemical transport model – technical description, Atmos. Chem. Phys., 12, 7825–7865, https://doi.org/10.5194/acp-12-7825-2012, 2012. a, b, c, d, e, f, g, h, i
Simpson, D., Tsyro, S., and Wind, P.: Updates to the EMEP/MSC-W model, in: Transboundary particulate matter, photo-oxidants, acidifying and eutrophying components, EMEP Status Report 1/2015, The Norwegian Meteorological Institute, Oslo, Norway, 129–138, 2015. a
Simpson, D., Bergström, R., Tsyro, S., and Wind, P.: Updates to the EMEP/MSC-W model, 2019–2020, in: Transboundary particulate matter, photo-oxidants, acidifying and eutrophying components, EMEP Status Report 1/2020, The Norwegian Meteorological Institute, Oslo, Norway, 155–165, 2020a. a
Simpson, D., Bergström, R., Briolat, A., Imhof, H., Johansson, J., Priestley, M., and Valdebenito, A.: metno/genchem: GenChem v1.0, Zenodo, https://doi.org/10.5281/zenodo.4133566, 2020b. a
Solberg, S., Hov, O., Sovde, A., Isaksen, I. S. A., Coddeville, P., De Backer, H., Forster, C., Orsolini, Y., and Uhse, K.: European surface ozone in the extreme summer 2003, J. Geophys. Res., 113, D07307, https://doi.org/10.1029/2007JD009098, 2008. a
Squire, O. J., Archibald, A. T., Griffiths, P. T., Jenkin, M. E., Smith, D., and Pyle, J. A.: Influence of isoprene chemical mechanism on modelled changes in tropospheric ozone due to climate and land use over the 21st century, Atmos. Chem. Phys., 15, 5123–5143, https://doi.org/10.5194/acp-15-5123-2015, 2015. a, b
Stadtler, S., Simpson, D., Schröder, S., Taraborrelli, D., Bott, A., and Schultz, M.: Ozone impacts of gas–aerosol uptake in global chemistry transport models, Atmos. Chem. Phys., 18, 3147–3171, https://doi.org/10.5194/acp-18-3147-2018, 2018. a
Stockwell, W., Kirchner, F., and Kuhn, M.: A New Mechanism for Regional Atmospheric Chemistry Modeling, J. Geophys. Res., 102, 25847–25879, 1997. a
Stroud, C. A., Zaganescu, C., Chen, J., McLinden, C. A., Zhang, J., and Wang, D.: Toxic volatile organic air pollutants across Canada: multi-year concentration trends, regional air quality modelling and source apportionment, J. Atmos. Chem., 73, 137–164, https://doi.org/10.1007/s10874-015-9319-z, 2016. a
Surendran, D. E., Ghude, S. D., Beig, G., Emmons, L., Jena, C., Kumar, R., Pfister, G., and Chate, D.: Air quality simulation over South Asia using Hemispheric Transport of Air Pollution version-2 (HTAP-v2) emission inventory and Model for Ozone and Related chemical Tracers (MOZART-4), Atmos. Environ., 122, 357–372, https://doi.org/10.1016/j.atmosenv.2015.08.023, 2015. a
Topping, D., Connolly, P., and Reid, J.: PyBox: An automated box-model generator for atmospheric chemistry and aerosol simulations., J. Open Source Software, 3, 755, https://doi.org/10.21105/joss.00755, 2018. a
Tsyro, S., Simpson, D., Tarrasón, L., Klimont, Z., Kupiainen, K., Pio, C., and Yttri, K. E.: Modeling of elemental carbon over Europe, J. Geophys. Res., 112, D23S19, https://doi.org/10.1029/2006JD008164, 2007. a
Verwer, J. G.: Gauss-Seidel iterations for stiff ODEs from chemical kinetics, SIAM J. Sci. Comput., 15, 1243–1250, https://doi.org/10.1137/0915076, 1994. a
Verwer, J. and Simpson, D.: Explicit methods for stiff ODEs from atmospheric chemistry, Appl. Numer. Math., 18, 413–430, 1995. a
Verwer, J. G., Blom, J. G., Van Loon, M., and Spee, E. J.: A comparison of stiff ODE solvers for atmospheric chemistry problems, Atmos. Environ., 30, 49–58, 1996. a
Vieno, M., Dore, A. J., Bealey, W. J., Stevenson, D. S., and Sutton, M. A.: The importance of source configuration in quantifying footprints of regional atmospheric sulphur deposition, Science of the Total Environment, 408, 985–995, https://doi.org/10.1016/j.scitotenv.2009.10.048, 2010. a
Vieno, M., Heal, M. R., Twigg, M. M., MacKenzie, I. A., Braban, C. F., Lingard, J. J. N., Ritchie, S., Beck, R. C., Móring, A., Ots, R., Marco, C. F. D., Nemitz, E., Sutton, M. A., and Reis, S.: The UK particulate matter air pollution episode of March–April 2014: more than Saharan dust, Environ. Res. Lett., 11, 044004, https://doi.org/10.1088/1748-9326/11/4/044004, 2016. a
Watson, L. A., Shallcross, D. E., Utembe, S. R., and Jenkin, M. E.: A Common Representative Intermediates (CRI) mechanism for VOC degradation. Part 2: Gas phase mechanism reduction, Atmos. Environ., 42, 7196–7204, https://doi.org/10.1016/j.atmosenv.2008.07.034, 2008. a
Yarwood, G., Jung, J., Heo, G., Whitten, G. Z., Mellberg, J., and Estes, M.: CB6 Version 6 of the Carbon Bond Mechanism, in: 9th Annual CMAS Conference, Chapel Hill, North Carolina, 11–13 October 2010, available at: https://www.cmascenter.org/conference/2010/abstracts/emery_updates_carbon_2010.pdf (last access: 2 December 2020), 2010a. a
Yarwood, G., Whitten, G. Z., Jung, J., Heo, G., and Allen, D. T.: Development, Evaluation and Testing of Version 6 of the Carbon Bond Chemical Mechanism (CB6), Final report to the Texas Commision on Environmental Quality, 582-7-84005-FY10-26, ENVIRON International Corporation, available at: http://www.tceq.texas.gov/assets/public/implementation/air/am/contracts/reports/pm/5820784005FY1026-20100922-environ-cb6.pdf (last access: last access: 2 December 2020), 2010b. a
- Abstract
- Introduction
- Installation and code
- Getting started – GenChem and boxChem basics
- Generating Fortran code for the EMEP CTM model
- Chemical mechanisms in GenChem
- Defining chemical mechanisms
- GenChem outputs: Fortran modules and included files
- Conclusions
- Code availability
- Author contributions
- Competing interests
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement
- Abstract
- Introduction
- Installation and code
- Getting started – GenChem and boxChem basics
- Generating Fortran code for the EMEP CTM model
- Chemical mechanisms in GenChem
- Defining chemical mechanisms
- GenChem outputs: Fortran modules and included files
- Conclusions
- Code availability
- Author contributions
- Competing interests
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement