the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 20 Nov 2020
| 20 Nov 2020
Oceanic and atmospheric methane cycling in the cGENIE Earth system model – release v0.9.14
Christopher T. Reinhard
Stephanie L. Olson
Sandra Kirtland Turner
Cecily Pälike
Yoshiki Kanzaki
Andy Ridgwell
The methane (CH4) cycle is a key component of the Earth system that links planetary climate, biological metabolism, and the global biogeochemical cycles of carbon, oxygen, sulfur, and hydrogen. However, currently lacking is a numerical model capable of simulating a diversity of environments in the ocean, where CH4 can be produced and destroyed, and with the flexibility to be able to explore not only relatively recent perturbations to Earth's CH4 cycle but also to probe CH4 cycling and associated climate impacts under the very low-O2 conditions characteristic of most of Earth's history and likely widespread on other Earth-like planets. Here, we present a refinement and expansion of the ocean–atmosphere CH4 cycle in the intermediate-complexity Earth system model cGENIE, including parameterized atmospheric O2–O3–CH4 photochemistry and schemes for microbial methanogenesis, aerobic methanotrophy, and anaerobic oxidation of methane (AOM). We describe the model framework, compare model parameterizations against modern observations, and illustrate the flexibility of the model through a series of example simulations. Though we make no attempt to rigorously tune default model parameters, we find that simulated atmospheric CH4 levels and marine dissolved CH4 distributions are generally in good agreement with empirical constraints for the modern and recent Earth. Finally, we illustrate the model's utility in understanding the time-dependent behavior of the CH4 cycle resulting from transient carbon injection into the atmosphere, and we present model ensembles that examine the effects of atmospheric pO2, oceanic dissolved SO, and the thermodynamics of microbial metabolism on steady-state atmospheric CH4 abundance. Future model developments will address the sources and sinks of CH4 associated with the terrestrial biosphere and marine CH4 gas hydrates, both of which will be essential for comprehensive treatment of Earth's CH4 cycle during geologically recent time periods.
- Article
(3778 KB) - Full-text XML
-
Supplement
(92 KB) - BibTeX
- EndNote





The global biogeochemical cycle of methane (CH4) is central to the evolution and climatic stability of the Earth system. Methane provides an important substrate for microbial metabolism, particularly in energy-limited microbial ecosystems in the deep subsurface (Valentine, 2011; Chapelle et al., 1995) and in anoxic marine and lacustrine sediments (Lovley et al., 1982; Hoehler et al., 2001). Indeed, the microbial production and consumption of CH4 are amongst the oldest metabolisms on Earth, with an isotopic record of bacterial methane cycling stretching back nearly 3.5 billion years (Ueno et al., 2006; Hinrichs, 2002; Hayes, 1994). As the most abundant hydrocarbon in Earth's atmosphere CH4 also has a significant influence on atmospheric photochemistry (Thompson and Cicerone, 1986), and because it absorbs in a window region of Earth's longwave emission spectrum it is an important greenhouse gas. This has important implications over the coming centuries, with atmospheric CH4 classified as a critical near-term climate forcing (Myhre et al., 2013), but has also resulted in dramatic impacts during certain periods of Earth's history. For example, high steady-state atmospheric CH4 has been invoked as an important component of Earth's early energy budget, potentially helping to offset a dim early Sun (Sagan and Mullen, 1972; Pavlov et al., 2000; Haqq-Misra et al., 2008), while time-dependent changes to the atmospheric CH4 inventory have been invoked as drivers of extreme climatic perturbations throughout Earth's history (Dickens et al., 1997; Dickens, 2003; Bjerrum and Canfield, 2011; Zeebe, 2013; Schrag et al., 2002). Because it is cycled largely through biological processes on the modern (and ancient) Earth and is spectrally active, atmospheric CH4 has also been suggested as a remotely detectable biosignature that could be applied to planets beyond our solar system (Hitchcock and Lovelock, 1967; Sagan et al., 1993; Krissansen-Totton et al., 2018).
A number of low-order Earth system models incorporating a basic CH4 cycle have been developed, particularly with a view to addressing relatively “deep time” geological questions. These include explorations of long-term changes to the chemistry of Earth's atmosphere (Claire et al., 2006; Catling et al., 2007; Bartdorff et al., 2008; Beerling et al., 2009), potential climate impacts at steady state (Kasting et al., 2001; Ozaki et al., 2018), and transient impacts of CH4 degassing on climate (Schrag et al., 2002; Bjerrum and Canfield, 2011). In some cases these models explicitly couple surface fluxes to a model of atmospheric photochemistry (Lamarque et al., 2006; Ozaki et al., 2018; Kasting et al., 2001), but in general atmospheric chemistry is parameterized based on offline 1-D or 2-D photochemical models, while surface fluxes are specified arbitrarily or are based on a simple one-box ocean–biosphere model. A range of slightly more complex “box” model approaches have been applied to simulate transient perturbations to Earth's CH4 cycle and attendant climate impacts on timescales ranging from ∼ 105 years (Dickens et al., 1997; Dickens, 2003) to ∼ 108 years (Daines and Lenton, 2016). In addition, offline and/or highly parameterized approaches toward simulating the impact of transient CH4 degassing from gas hydrate reservoirs have been developed and applied to relatively recent periods of Earth's history (Archer and Buffett, 2005; Lunt et al., 2011) or projected future changes (Archer et al., 2009; Hunter et al., 2013). However, the most sophisticated and mechanistic models of global CH4 cycling currently available tend to focus on terrestrial (soil or wetland) sources and sinks (Ridgwell et al., 1999; Walter and Heimann, 2000; Wania et al., 2010; Konijnendijk et al., 2011; Melton et al., 2013) or on explicitly modeling atmospheric photochemistry (Shindell et al., 2013).
Much less work has been done to develop ocean biogeochemistry models that are both equipped to deal with the wide range of boundary conditions characteristic of Earth's history and are computationally tractable when running large model ensembles and/or on long (approaching ∼ 106 year) timescales, as well as being able to simulate the (3-D) redox structure of the ocean, allowing for localized zones of production and oxidation (which provides more accurate estimates of emission to the atmosphere). For instance, Elliot et al. (2011) advanced modeling of marine CH4 cycling by developing and employing a 3-D ocean circulation and climate model (CCSM-3) to simulate the impact of injecting clathrate-derived CH4 into the Arctic Ocean. However, microbial consumption of CH4 in the ocean interior was parameterized via an empirical log-linear function that implicitly neglects anaerobic oxidation of methane (AOM) via dissolved sulfate (SO), which on the modern Earth is an enormously important internal CH4 sink within Earth's oceans (Egger et al., 2018). Their simulations did not explore atmospheric chemistry. Similarly, Daines and Lenton (2016) also innovated over traditional box modeling approaches by applying an ocean general circulation model (GCM) to examine the role of aerobic methanotrophy in modulating ocean–atmosphere fluxes of CH4 during the Archean (prior to ∼ 2.5 billion years ago, Ga). However, this analysis likewise did not include AOM, and the GCM results were not coupled to atmospheric chemistry. In contrast, Olson et al. (2016) included AOM in a 3-D ocean biogeochemistry model coupled to an atmospheric chemistry routine and found that AOM represents a critical internal CH4 sink in the oceans even at relatively low dissolved SO levels. Though this represented an important further step forward in understanding marine CH4 cycling on the early Earth, Olson et al. (2016) employed a simplified parameterization of aerobic CH4 consumption, neglected the thermodynamics of CH4-consuming metabolisms under energy-limited conditions, and employed a parameterization of atmospheric O2–O3–CH4 photochemistry that is most readily applicable to only a subset of the atmospheric pO2 values characteristic of Earth's history (Daines and Lenton, 2016; Olson et al., 2016). While all of these studies provided new modeling innovations and advances in understanding, important facets of global CH4 cycling, particularly as relevant to the evolution of the early Earth, were lacking.
Here, we present a new framework for modeling the ocean–atmosphere biogeochemical CH4 cycle in the “muffin” release of the cGENIE Earth system model. Our goal is to make further progress in the development of a flexible intermediate-complexity model suitable for simulating the global biogeochemical CH4 cycle on ocean-bearing planets, with an initial focus on periods of Earth's history (or other habitable planets) that lack a robust terrestrial biosphere. We also aim to provide a numerical modeling foundation from which to further develop a more complete CH4 cycle within the cGENIE framework, including, for example, dynamic CH4 hydrate cycling and the production and consumption of CH4 by terrestrial ecosystems.
The outline of the paper is as follows. In Sect. 2 we briefly describe the GENIE–cGENIE Earth system model, with a particular eye toward the features that are most relevant for the biological carbon pump and the oceanic CH4 cycle. In Sect. 3 we describe the major microbial metabolisms involved in the oceanic CH4 cycle and compare our parameterizations to data from modern marine environments. In Sect. 4 we describe two alternative parameterizations of atmospheric O2–O3–CH4 photochemistry incorporated into the model and compare these to modern and more recent observations. In Sect. 5 we present results from a series of idealized simulations meant to illustrate the flexibility of the model and some potential applications. The availability of the model code, plus configuration files for all experiments described in the paper, is provided in Sect. 7, following a brief summary in Sect. 6.
2.1 Ocean physics and climate model – C-GOLDSTEIN
The ocean physics and climate model in cGENIE are comprised of a reduced physics (frictional geostrophic) 3-D ocean circulation model coupled to both a 2-D energy–moisture balance model (EMBM) and a dynamic–thermodynamic sea ice model (Edwards and Marsh, 2005; Marsh et al., 2011). The ocean model transports heat, salinity, and biogeochemical tracers using a scheme of parameterized isoneutral diffusion and eddy-induced advection (Griffies, 1998; Edwards and Marsh, 2005; Marsh et al., 2011), exchanges heat and moisture with the atmosphere, sea ice, and land, and is forced at the ocean surface by the input of zonal and meridional wind stress according to a specified wind field. The 2-D atmospheric energy–moisture balance model (EMBM) considers the heat and moisture balance for the atmospheric boundary layer using air temperature and specific humidity as prognostic tracers. Heat and moisture are mixed horizontally throughout the atmosphere and exchange heat and moisture with the ocean and land surfaces with precipitation occurring above a given relative humidity threshold. The sea ice model tracks the horizontal transport of sea ice and the exchange of heat and fresh water with the ocean and atmosphere using ice thickness, areal fraction, and concentration as prognostic variables. Full descriptions of the model and coupling procedure can be found in Edwards and Marsh (2005) and, more recently, in Marsh et al. (2011). As implemented here, the ocean model is configured as a 36×36 equal-area grid (uniform in longitude and uniform in the sine of latitude) with 16 logarithmically spaced depth levels and seasonal surface forcing from the EMBM.
2.2 Ocean biological pump – BIOGEM
The biogeochemical model component – BIOGEM – regulates air–sea gas exchange as well as the transformation and partitioning of biogeochemical tracers within the ocean, as described in Ridgwell et al. (2007). By default, the biological pump is driven by parameterized uptake of nutrients in the surface ocean, with this flux converted stoichiometrically to biomass that is then partitioned into either dissolved or particulate organic matter for downstream transport, sinking, and remineralization. Dissolved organic matter is transported by the ocean model and decays with a specified time constant, while particulate organic matter is immediately exported out of the surface ocean and partitioned into two fractions of differing lability. In the ocean interior, particulate organic matter is remineralized instantaneously throughout the water column following an exponential decay function with a specified remineralization length scale.
In the simulations discussed below, photosynthetic nutrient uptake in surface ocean grid cells is controlled by a single limiting nutrient, dissolved phosphate (PO4):
where DOP represents dissolved organic phosphorus, υ represents the proportion of photosynthetic production that is initially partitioned into a dissolved organic phase, λ represents a decay constant (time−1) for dissolved organic matter, and Γ represents photosynthetic nutrient uptake following Doney et al. (2006):
Rates of photosynthesis are regulated by terms describing the impact of available light (FI), nutrient abundance (FN), temperature (FT), and fractional sea ice coverage in each grid cell (fice). Rates of photosynthetic nutrient uptake are further scaled to ambient dissolved PO4 ([PO4]) according to an optimal uptake timescale (τbio).
Note that this parameterization differs from that in Ridgwell et al. (2007). Specifically, the impacts of light and nutrient availability are both described via Michaelis–Menten terms:
where shortwave irradiance I is averaged over the entire mixed layer and is assumed to decay exponentially from the sea surface with a length scale of 20 m. It is assumed that nutrient uptake and photosynthetic production only occur in surface grid cells of cGENIE (e.g., the upper 80 m), which is similar to the “compensation depth” zc in Doney et al. (2006) of 75 m. The terms κI and κP represent half-saturation constants for light and dissolved phosphate, respectively. In addition, the effect of temperature on nutrient uptake is parameterized according to
where kT0 and keT denote pre-exponential and exponential scaling constants and T represents absolute in situ temperature. The scaling constants are chosen to give approximately a factor of 2 change in rate with a temperature change of 10 ∘C (e.g., a Q10 response of ∼ 2.0). Lastly, the final term in Eq. (3), not present in the default parameterization of Ridgwell et al. (2007), allows biological productivity to scale more directly with available PO4 when dissolved PO4 concentrations are elevated relative to those of the modern oceans.
Particulate organic matter (POM) is immediately exported out of the surface ocean without lateral advection and is instantaneously remineralized throughout the water column according to an exponential function of depth:
where is the particulate organic matter flux at a given depth (and zh is the base of the photic zone), z is depth, and and refer to the relative partitioning into each organic matter lability fraction i and the e-folding depth of that fraction, respectively. The simulations presented here employ two organic matter fractions, a “labile” fraction (94.5 %) with an e-folding depth of ∼ 590 m and an effectively inaccessible fraction (5.5 %) with an e-folding depth of 106 m (Table 1).
Table 1Default parameters for organic matter production and water column remineralization.
1 Ridgwell et al. (2007); 2 Meyer et al. (2016); 3 Doney et al. (2006); 4 Olson et al. (2016).
We employ a revised scheme for organic matter remineralization in the ocean interior, following that commonly used in models of organic matter remineralization within marine and lacustrine sediments (Rabouille and Gaillard, 1991; Van Cappellen et al., 1993; Boudreau, 1996a, b). Respiratory electron acceptors (O2, NO, and SO) are consumed according to decreasing free energy yield (Froelich et al., 1979), with consumption rates (Ri) scaled to both electron acceptor abundance and the inhibitory impact of electron acceptors with higher intrinsic free energy yield:
with the exception that in the biogeochemical configuration used here we do not consider nitrate (NO). The total consumption of settling POM within each ocean layer is governed by the predetermined remineralization profiles (Eq. 7). The Ri terms denote the relative fraction of this organic matter consumption that is performed by each respiratory process. We specify a closed system with no net organic matter burial in marine sediments (see below), and hence the POM flux to the sediment surface is assumed to be completely degraded, with the same partitioning amongst electron acceptors carried out according to local bottom water chemistry. For DOM, the assumed lifetime (λ) determines the total fraction of DOM degraded (and Eqs. 8–10 again determine how the consumption of electron acceptors is partitioned). The κi terms represent half-saturation constants for each metabolism, the terms give inhibition constants acting on less energetic downstream respiratory processes, and brackets denote concentration. Default parameter values used here are shown in Table 1.
3.1 Microbial methanogenesis
Methanogenesis represents the terminal step in our remineralization scheme and follows the overall stoichiometry:
This can be taken to implicitly include fermentation of organic matter to acetate followed by acetoclastic methanogenesis:
or the fermentation of organic matter to acetate followed by anaerobic acetate oxidation and hydrogenotrophic methanogenesis:
both of which have the same overall net stoichiometry provided that H2 is assumed to be quantitatively converted to CH4 by hydrogenotrophic methanogens. We thus ignore the scenario in which some fraction of H2 is converted directly to biomass by hydrogenotrophic methanogens acting as primary producers (Ozaki et al., 2018).
Because we specify a closed system with no net organic matter burial in marine sediments, all organic matter not remineralized by more energetic respiratory metabolisms is converted into CH4 (e.g., ):
where the κi and terms are as described above (Table 1). We disable nitrate (NO3) as a tracer in the simulations presented here such that anaerobic remineralization of organic matter is partitioned entirely between sulfate reduction and methanogenesis (Fig. 1). Using our default parameter values (Table 1), aerobic respiration dominates organic matter remineralization at [O2] values significantly above 1 µmol kg−1 (Fig. 1a), while anaerobic remineralization is dominated by methanogenesis at [SO] values significantly below 1 mmol kg−1 (Fig. 1b). An important outcome of the revised “inhibition” scheme is that metabolic pathways with differing intrinsic free energy yields can coexist, which more accurately reflects field observations from a range of natural settings (Curtis, 2003; Bethke et al., 2008; Kuivila et al., 1989; Jakobsen and Postma, 1999). In particular, it allows us to roughly capture the impact of oxidant gradients within sinking marine aggregates (Bianchi et al., 2018), which can facilitate nontrivial anaerobic carbon remineralization within sinking particles even in the presence of relatively high [O2] in the ocean water column (Fig. 1c).
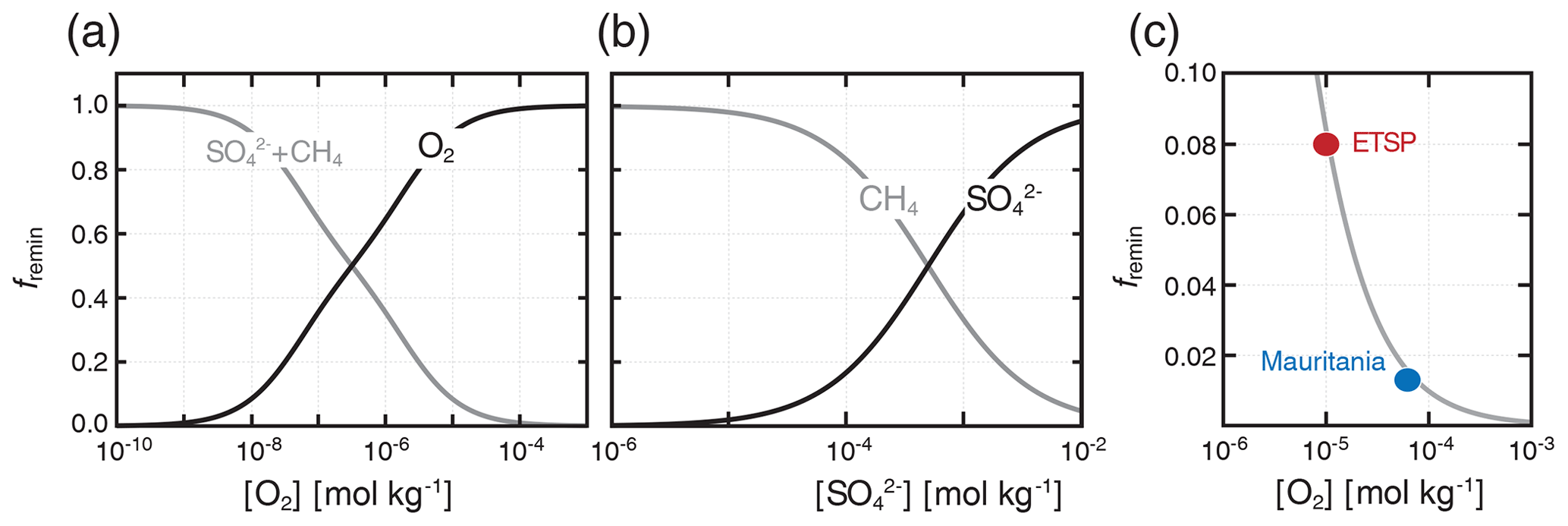
Figure 1Fractional organic carbon remineralization by aerobic respiration, sulfate reduction, and methanogenesis in our modified organic matter remineralization scheme. In (a), relative rates of aerobic (O2) and anaerobic (SO CH4) remineralization are plotted as a function of dissolved [O2]. In (b), relative anaerobic remineralization rates are partitioned between sulfate reduction and methanogenesis as a function of dissolved [SO] (dissolved [O2] is held constant at 10−10 mol kg−1). Shown in (c) are our estimated anaerobic remineralization fractions (grey curve) compared to estimates from a particle biogeochemical model applied to oxygen minimum zones (OMZs) in the eastern tropical South Pacific (ETSP) and Mauritanian upwelling (Bianchi et al., 2018).
While the model tracks the carbon isotope composition of oceanic and atmospheric CH4 (δ13C, reported in per mill notation relative to the Pee Dee Belemnite, PDB), the only significant isotope effect we include here is that attendant to acetoclastic methanogenesis. We specify a constant isotope fractionation between organic carbon and CH4 during methanogenesis of −35 ‰ by default (Table 2), which will tend to produce microbial CH4 with a δ13C composition of roughly −60 ‰ when combined with the default isotope fractionation associated with photosynthetic carbon fixation in the surface ocean (e.g., Kirtland Turner and Ridgwell, 2016). The model does not currently include any potential isotope effects associated with aerobic and anaerobic methanotrophy, air–sea gas exchange of CH4, or photochemical breakdown of CH4 in the atmosphere. It does, however, include a comprehensive 13C scheme associated with ocean–atmosphere cycling of CO2 (Kirtland Turner and Ridgwell, 2016; Ridgwell, 2001).
Table 2Default kinetic and thermodynamic parameters for oceanic methane cycling. Activity coefficients are estimated for T=25 ∘C and S=35 ‰.
1 Olson et al. (2016); 2 Regnier et al. (2011); 3 Schink (1997); 4 Hoehler et al. (2001); 5 Hoehler (2004); 6 Stoessell and Byrne (1982); 7 Cramer (1984); 8 Duan et al. (1992); 9 Johnson (1982); 10 Clegg and Brimblecombe (1990); 11 Ulfsbo et al. (2015); 12 Berner (1965); 13 Helz et al. (2011); 14 Dale et al. (2008).
3.2 Aerobic methanotrophy
Microbial aerobic methanotrophy proceeds according to
This reaction is highly favorable energetically, with a free energy yield under standard conditions of ∼ 850 kJ per mole of methane consumed (Table 2). We represent rates of aerobic methanotrophy (RAER) with a mixed kinetic–thermodynamic formulation (Jin and Bethke, 2005, 2007; Regnier et al., 2011), in which CH4 oxidation kinetics are controlled by substrate availability, thermodynamic energy yield, and temperature:
A rate constant for aerobic methanotrophy (yr−1) is defined as kAER, while Fi terms denote kinetic (k) and thermodynamic (t) factors as defined below and a temperature (T) factor as given in Eq. (6) above.
The kinetic factor (Fk) for aerobic methanotrophy is controlled by substrate availability according to
where brackets denote concentration and the κ term denotes a half-saturation constant with respect to O2. We employ a hybrid parameterization in which kinetics are first-order with respect to CH4 but also scaled by a Michaelis–Menten-type term for O2. This formulation is based on the rationale that half-saturation constants for CH4 are typically similar to (or greater than) the dissolved CH4 levels attained in anoxic water column environments (Regnier et al., 2011) but is also meant to allow for rapid CH4 consumption under “bloom” conditions with an appropriately scaled rate constant (see below).
The effect of thermodynamic energy yield on aerobic methanotrophy is given by
where ΔGr denotes the Gibbs free energy of reaction under in situ conditions, ΔGBQ represents the minimum energy required to sustain ATP synthesis (Hoehler et al., 2001; Hoehler, 2004; Jin and Bethke, 2007), χ is the stoichiometric number of the reaction (e.g., the number of times the rate-determining step occurs in the overall process), and R and T represent the gas constant and absolute in situ temperature, respectively. The available free energy is estimated according to
where, in addition to the terms defined above, represents the Gibbs free energy of the reaction under standard conditions, and γi values represent activity coefficients. Note that we assume an H2O activity of unity.
3.3 Anaerobic oxidation of methane (AOM)
The oxidation of methane can also be coupled to electron acceptors other than O2, including nitrate (NO), sulfate (SO), and oxide phases of iron (Fe) and manganese (Mn) (Reeburgh, 1976; Martens and Berner, 1977; Hoehler et al., 1994; Hinrichs et al., 1999; Orphan et al., 2001; Sivan et al., 2011; Haroon et al., 2013; Egger et al., 2015). Because it is by far the most abundant of these oxidants on the modern Earth and has likely been the most abundant throughout Earth's history, we focus on anaerobic oxidation of methane (AOM) at the expense of SO:
This process is currently thought to be performed most often through a syntrophic association between Archaea and sulfate-reducing bacteria (Boetius et al., 2000), though the mechanics controlling the exchange of reducing equivalents within the syntrophy remain to be fully elucidated (Milucka et al., 2012; McGlynn et al., 2015). In any case, consumption of CH4 at the sulfate–methane transition zone (SMTZ) represents an extremely large sink flux of CH4 in modern marine sediments (Regnier et al., 2011; Egger et al., 2018).
Anaerobic methanotrophy is much less energetically favorable under standard conditions, with a free energy yield of ∼ 30 kJ per mole of CH4 (Table 2). As a result, the influence of thermodynamics on rates of AOM is potentially much stronger than it will tend to be in the case of aerobic methanotrophy. As above, rates of AOM are controlled by the combined
where kAOM is a rate constant for anaerobic methane oxidation (yr−1), while Fi terms denote kinetic (k) and thermodynamic (t) factors as defined below and a temperature (T) factor as given in Eq. (6) above.
The kinetics of anaerobic methane oxidation are specified according to
where brackets denote concentration and the κ term denotes a half-saturation constant with respect to SO. We employ a hybrid parameterization in which kinetics are first-order with respect to CH4 but are also scaled by a Michaelis–Menten-type term for SO for reasons discussed above.
The effect of thermodynamic energy yield on anaerobic methane oxidation is specified as follows:
As above, ΔGr denotes the Gibbs free energy of reaction under in situ conditions, ΔGBQ is the minimum energy required to sustain ATP synthesis (the “biological quantum”), χ is the stoichiometric number of the reaction, and R and T represent the gas constant and absolute in situ temperature, respectively. The available free energy for AOM under in situ conditions is estimated according to
where again represents the Gibbs free energy of the net AOM reaction given above under standard conditions, and γi values represent activity coefficients. Again, we assume an H2O activity of unity.
3.4 Default parameters for aerobic and anaerobic methanotrophy
We choose default rate constants according to a dataset of compiled rates of aerobic and anaerobic methanotrophy in oxygenated and anoxic marine water column environments (see the Supplement), after correction to in situ temperature (Fig. 2a, b). Our default values for both rate constants are on the low end of the observational dataset but are very roughly tuned to yield steady-state diffusive CH4 fluxes from the ocean that are consistent with recent observational constraints (Fig. 2c). It is important to note, however, that these values are not extensively tuned and could be adjusted depending on the application. For example, transient CH4 release experiments could employ rate constants that are scaled upward to reflect transient (bloom) elevations in microbial community CH4 consumption as observed in field studies (Kessler et al., 2011; Crespo-Medina et al., 2014). Default values for other kinetic parameters (Table 2) are chosen to be broadly consistent with field measurements and pure or mixed culture experiments with aerobic methanotrophs (Bender and Conrad, 1992, 1993; Hanson and Hanson, 1996; Dunfield and Conrad, 2000; van Bodegom et al., 2001), as well as to remain roughly consistent with previous work for comparative purposes (e.g., Olson et al., 2016), though the parameters have not been formally tuned and we explore model sensitivity below.

Figure 2Compilation of rate constants for aerobic (AER; a) and anaerobic (AOM; b) methane oxidation. Rate constants are corrected for in situ temperature using a Q10 of 2 (see the Supplement). Vertical red lines show our default values as reported in Table 2. Shown in (c) are globally integrated diffusive fluxes of CH4 from the ocean for a range of rate constants for aerobic methanotrophy, including our default simulation. The bar to the right of (c) shows the median (black bar) and 90 % credible interval (grey shading) for estimates of the modern oceanic diffusive flux from Weber et al. (2019).
Thermodynamic energy yields of each reaction under standard conditions are calculated based on the standard molal thermodynamic properties given in Regnier et al. (2011). Stoichiometric numbers are assumed identical for both metabolisms, with default values of 1.0 (Jin and Bethke, 2005; Dale et al., 2006). We assume a default biological quantum (ΔGBQ) of 15 kJ mol−1 for both aerobic and anaerobic methanotrophy, though these can be expected to vary somewhat as a function of metabolism and environmental conditions (Schink, 1997; Hoehler, 2004; Dale et al., 2008). These can be varied independently for aerobic and anaerobic methanotrophy in the model, and we explore model sensitivity to this parameter below. Lastly, for simplicity and to minimize computational expense we assume constant activity coefficients for each species throughout all ocean grid cells (Table 2). For some applications it may ultimately be important to add a scheme for estimating activity coefficients according to ambient salinity and ion chemistry, for example estimating methane fluxes in planetary scenarios with very different major ion chemistry or much higher or lower salinity than those characteristic of Earth's modern oceans.
4.1 Air–sea gas exchange
Ocean–atmosphere fluxes of CH4 (Jgas) are governed by temperature- and salinity-dependent solubility and surface wind speed above a given grid cell:
where A denotes the area available for gas exchange (e.g., the area of ice-free surface ocean), [CH4]cell denotes the ambient dissolved CH4 concentration in a given surface ocean grid cell, [CH4]sat represents the dissolved CH4 concentration at saturation with a given atmospheric pCH4, temperature, and salinity, and kgas represents a gas transfer velocity. Solubility is based on a Bunsen solubility coefficient (β) corrected for ambient temperature (T) and salinity (S) according to
Note that the Henry's law constant K0 is related to the Bunsen solubility coefficient by , where ρ is density and V+ is the molar volume of the gas at standard temperature and pressure (STP). Gas transfer velocity (kgas) is calculated based on the surface wind speed (u) and a Schmidt number (Sc) corrected for temperature assuming a constant salinity of 35 ‰:
where k is a dimensionless gas transfer coefficient, u is surface wind speed, and Sc is the temperature-corrected Schmidt number according to
All default constants and coefficients for the gas exchange scheme are given in Table 3. Overall, the scheme for air–sea gas exchange of CH4 follows by default that for other gases accounted for in BIOGEM, such as O2 and CO2, as described in Ridgwell et al. (2007)
Table 3Default constants and coefficients for CH4 gas exchange. All default parameter values are derived from Wanninkhof (1992). Schmidt number coefficients are for S=35 ‰.
4.2 Parameterized O2–O3–CH4 photochemistry
Once degassed to the atmosphere, CH4 becomes involved in a complex series of photochemical reactions initiated by hydroxyl radical (OH) attack on CH4 (Kasting et al., 1983; Prather, 1996; Pavlov et al., 2000; Schmidt and Shindell, 2003). Following Claire et al. (2006) and Goldblatt et al. (2006), we parameterize O2–O3–CH4 photochemistry according to a bimolecular “rate law”:
where Mi terms represent the atmospheric inventories of O2 and CH4, respectively, and keff denotes an effective rate constant (Tmol−1 yr−1) that is itself a complicated function of atmospheric O2, CH4, and CO2 (Claire et al., 2006). We note that in this parameterization, O3 abundance is not calculated explicitly, but rather the photochemical destruction rate of CH4 in the atmosphere is controlled by the combined atmospheric chemistry implicitly embedded within keff (Goldblatt et al., 2006; Claire et al., 2006).
At each time step, the distribution of chemical species (e.g., other than temperature and humidity) in the atmosphere is homogenized (Ridgwell et al., 2007) and keff is estimated based on the resulting instantaneous mean partial pressures of O2 and CH4 according to a bivariate fit to a large suite of 1-D atmospheric photochemical models. These photochemical model results (Mark Claire, personal communication, 2016) are derived following Claire et al. (2006). Briefly, values for keff are computed by a 1-D model of atmospheric photochemistry assuming a range of fixed surface mixing ratios of O2 and CH4 and a constant atmospheric CO2 of 10−2 bar. We then fit a fifth-order polynomial surface to these keff values as a function of atmospheric pO2 and pCH4 (Fig. 3).
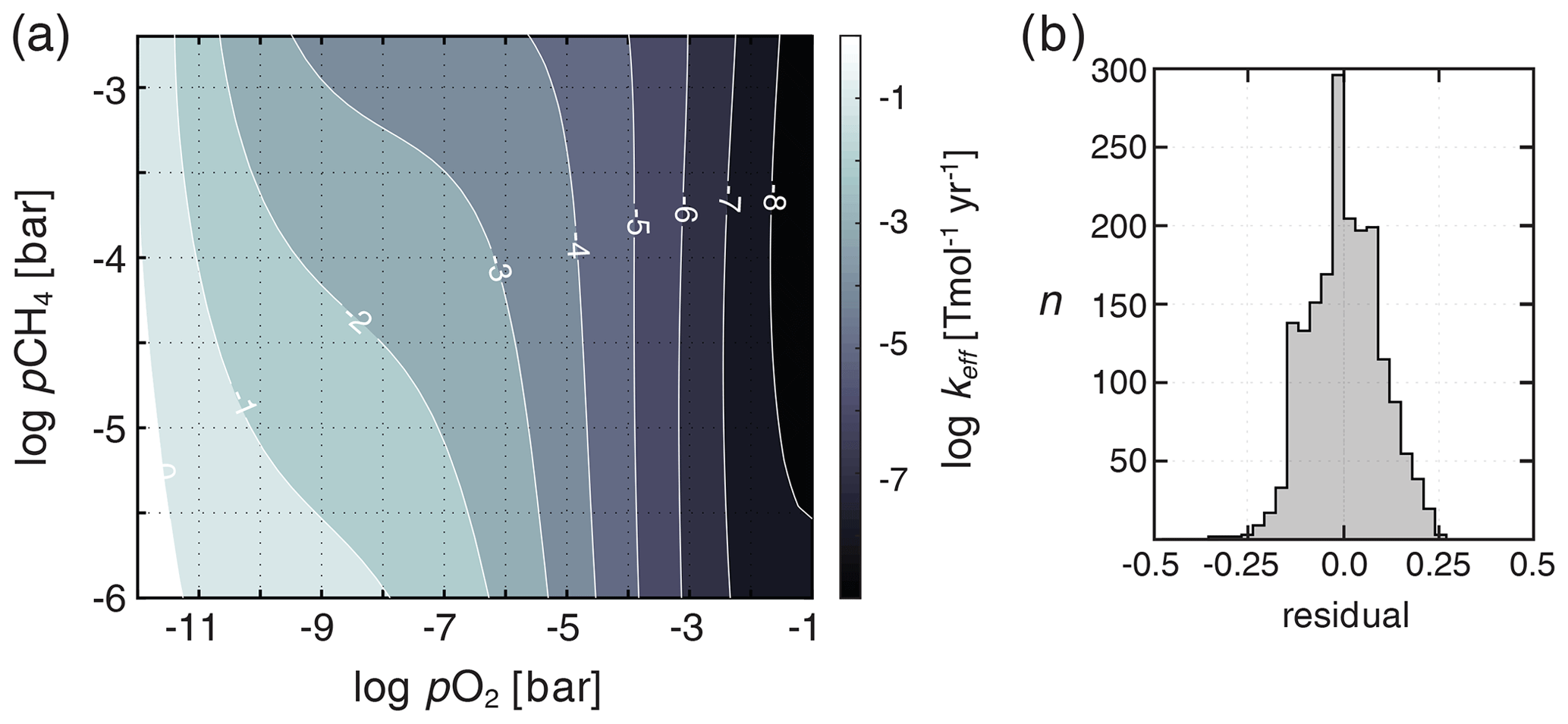
Figure 3Shown in (a) is the bivariate fit to a suite of 1-D atmospheric photochemical runs for the effective rate constant (keff) parameterizing O2–O3–CH4 photochemistry in ATCHEM. Shown in (b) is a frequency distribution of the residuals on keff from the underlying photochemical model output.
Our default parameterization of O2–O3–CH4 chemistry (C06) is fit over a pO2 range of 10−14 to 10−1 bar, a pCH4 range of 10−6 to bar, and a constant high background pCO2 of 10−2 bar (Claire et al., 2006). We thus truncate the atmospheric lifetime of CH4 at a lower bound of 7.6 years in our default parametrization and provide an alternative parameterization of photochemical CH4 destruction at roughly modern pO2 and pCO2 (SS03) derived from the results of Schmidt and Shindell (2003) for use in more geologically recent, high-O2 atmospheres (Reinhard et al., 2017) (Fig. 4a). Although this parameterized photochemistry scheme should represent an improvement in accuracy relative to that implemented in Olson et al. (2016) (see Daines and Lenton, 2016), it is important to point out that a range of factors that might be expected to impact the photochemical destruction rates of CH4 in the atmosphere, including atmospheric pCO2, the atmospheric profile of H2O, and spectral energy distribution (SED), have not yet been rigorously assessed. Ongoing model developments in ATCHEM are aimed at implementing a more flexible and inclusive photochemical parameterization that will allow for robust use across a wider range of atmospheric compositions and photochemical environments.
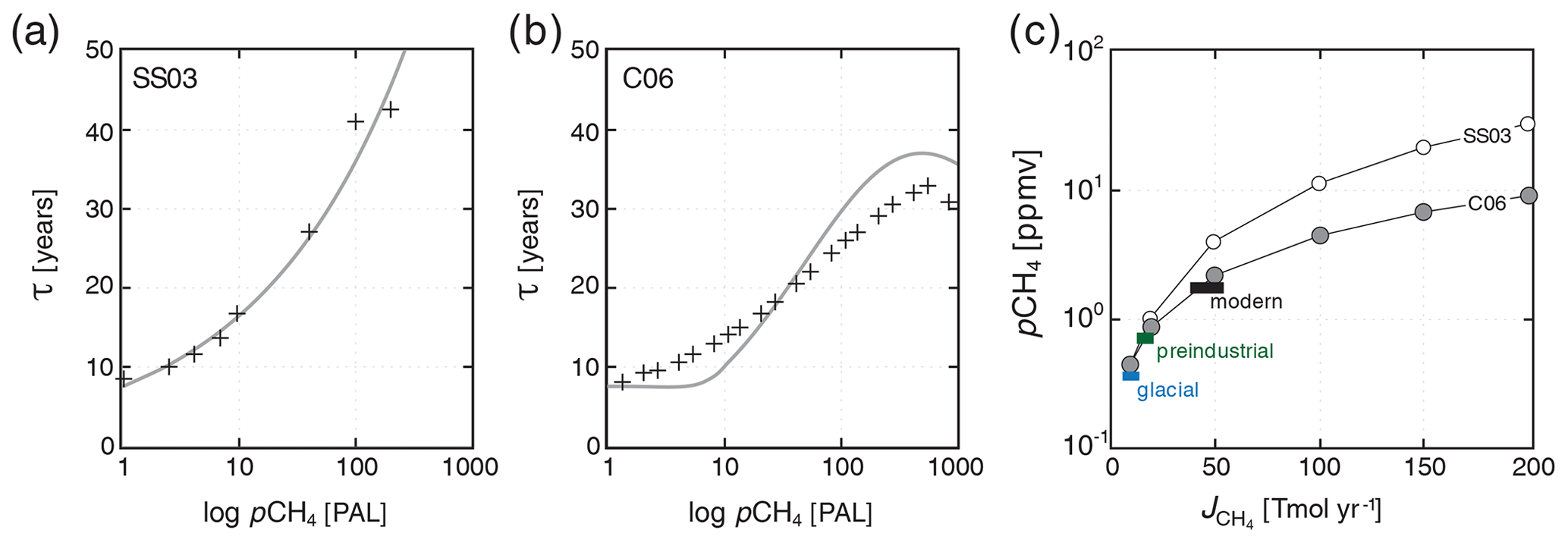
Figure 4Comparison of steady-state atmospheric pCH4 as a function of terrestrial CH4 flux with modern and more recent estimates. Shown in (a) is an exponential fit to the 2-D photochemistry model of Schmidt and Shindell (2003) (SS03), with individual model runs shown as black crosses. Shown in (b) is a plane through the bivariate fit shown in Fig. 3 (grey curve) compared with the ensemble of 1-D atmospheric photochemical models at pO2=0.1 atm (black crosses; see text). Shown in (c) are steady-state atmospheric CH4 values as a function of imposed terrestrial CH4 flux in our “modern” configuration (circles) compared to estimates for the glacial, preindustrial, and modern CH4 cycles (Kirschke et al., 2013; Bock et al., 2017; Paudel et al., 2016).
As a basic test of our photochemical parameterization, we impose a terrestrial (wetland) flux of CH4 to the atmosphere (balanced by stoichiometric consumption of CO2 and release of O2) and allow the oceanic and atmospheric CH4 cycle to spin up for 20 kyr. We then compare steady-state atmospheric pCH4 as a function of terrestrial CH4 flux to estimates for the last glacial, preindustrial, and modern periods. Our default parameterization is relatively simple and spans a very wide range in atmospheric O2 and CH4 inventories. Nevertheless, both the default scheme and the alternative parameterization for recent geologic history (and analogous planetary environments), with high-pO2,low-pCO2 atmospheres, accurately reproduce atmospheric pCH4 values given estimated glacial, preindustrial, and modern terrestrial CH4 fluxes (Fig. 4c), and both display the predicted saturation of CH4 sinks at elevated atmospheric CH4 observed in more complex photochemical models. We note, however, that the alternative parameterization tends to yield slightly higher atmospheric pCH4 at surface fluxes greater than ∼ 50 Tmol yr−1 (Fig. 4c). (In the remainder of the paper, we employ the default (C06) parameterization for atmospheric O2–O3–CH4 chemistry and do not discuss the simple high-pO2−low-pCO2 alternative further.)
5.1 High-pO2 (“modern”) steady state
We explore a roughly modern steady state with appropriate continental geography and simulated overturning circulation (as in Cao et al., 2009) and initialize the atmosphere with pO2, pCO2, and pCH4 of [v∕v] 20.95 %, 278 ppm, and 700 ppb, respectively, and with globally uniform oceanic concentrations of SO (28 mmol kg−1) and CH4 (1 nmol kg−1). We fix globally averaged solar insolation at the modern value (1368 W m−2) with seasonally variable forcing as a function of latitude and set radiative forcing for CO2 and CH4 equivalent to preindustrial values in order to isolate the effects of biogeochemistry on steady-state tracer distributions. The model is then spun up for 20 kyr with atmospheric pO2 and pCO2 (and δ13C of atmospheric CO2) restored to preindustrial values at every time step and with an imposed wetland flux of CH4 to the atmosphere of 20 Tmol yr−1 that has a δ13C value of −60 ‰. Atmospheric pCH4 and all oceanic tracers are allowed to evolve freely.
Surface, benthic, and ocean interior distributions of dissolved oxygen (O2), sulfate (SO), and methane (CH4) are shown in Fig. 5 for our roughly modern simulation. Dissolved O2 ([O2]) approaches air saturation throughout the surface ocean, with a distribution that is largely uniform zonally and with concentrations that increase with latitude as a result of increased solubility at lower temperature near the poles (Fig. 5a). Benthic [O2] shows patterns similar to those expected for the modern Earth, with relatively high values in the well-ventilated deep North Atlantic, low values in the deep North Pacific and Indian oceans, and a gradient roughly between air saturation near regions of deep convection in the high-latitude Atlantic and much lower values in the tropical and northern Pacific (Fig. 5d). Distributions of [O2] in the ocean interior are similar to those of the modern Earth (Fig. 5g) with oxygen minimum zones (OMZs) at intermediate depths underlying highly productive surface waters, particularly in association with coastal upwelling at low latitudes.
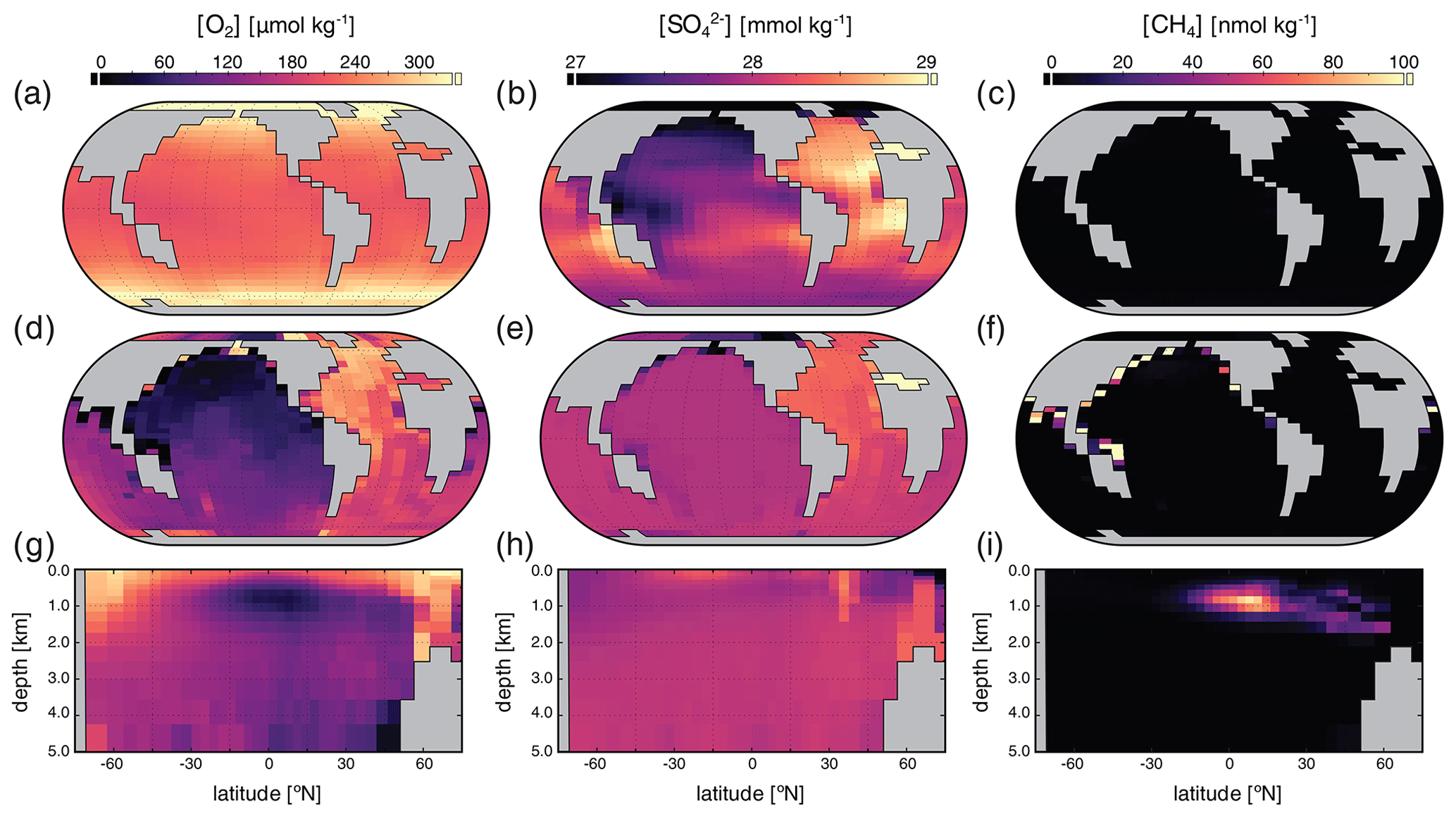
Figure 5Tracer distributions in surface (a–c) and benthic (d–f) grid cells and in the zonally averaged ocean interior (g–i) for O2 (a, d, g), SO (b, e, h), and CH4 (c, f, i) in our “modern” configuration. Note the different concentration units for each tracer.
Concentrations of dissolved SO ([SO]) are largely invariant throughout the ocean, consistent with its expected conservative behavior in the modern ocean as one of the most abundant negative ions in seawater (Fig. 5). Slightly higher concentrations in both surface and benthic fields are seen in association with outflow from the Mediterranean and are driven by evaporative concentration (Fig. 5b). Benthic [SO] distributions show some similarity to those of [O2] (Fig. 5e), though again the differences are very small relative to the overall prescribed initial tracer inventory of 28 mmol kg−1 and disappear almost entirely when salinity-normalized (not shown). In the ocean interior, [SO] is largely spatially invariant with a value of approximately 28 mmol kg−1 (Fig. 5h).
Dissolved CH4 concentrations ([CH4]) in the surface and shallow subsurface ocean are much more variable but typically on the order of ∼ 1–2 nmol kg−1, with slightly elevated concentrations just below the surface, both of which are consistent with observations from the modern ocean (Reeburgh, 2007; Scranton and Brewer, 1978). The benthic [CH4] distribution shows locally elevated values up to ∼ 300–400 nmol kg−1 in shallow regions of the tropical and northern Pacific and the Indian oceans (Fig. 5f), which is also broadly consistent with observations from shallow marine environments with active benthic CH4 cycling (Jayakumar et al., 2001). Within the ocean interior, dissolved CH4 can accumulate in the water column in excess of ∼ 100 nmol kg−1 in association with relatively low-O2 conditions at intermediate depths, with zonally averaged values as high as ∼ 70 nmol kg−1 but more typically in the range of ∼ 20–40 nmol kg−1 (Fig. 5i). These concentrations are comparable to those observed locally in low-O2 regions of the modern ocean (Sansone et al., 2001; Chronopoulou et al., 2017; Thamdrup et al., 2019).
The major metabolic fluxes within the ocean's microbial CH4 cycle for our “modern” configuration are shown in Fig. 6. Methanogenesis is focused in regions characterized by relatively low [O2] and is particularly vigorous in the eastern tropical Pacific, the North Pacific, and the Indian Ocean (Fig. 6a). The highest zonally averaged rates of methanogenesis are observed in northern tropical and subtropical latitudes and are focused at a depth of ∼ 1 km (Fig. 6d). Rates of microbial CH4 consumption are generally spatially coupled to rates of methanogenesis, both in a column-integrated sense (Fig. 6b, c) and in the zonal average (Fig. 6e, f). This is particularly true for AOM, the rates of which are highest within the core of elevated methanogenesis rates observed in the northern subtropics. Zonally averaged AOM rates of ∼ 10–15 nmol kg−1 d−1 compare well with field measurements of AOM within oceanic OMZs (Thamdrup et al., 2019). In general, the bulk of CH4 produced via microbial methanogenesis is consumed via AOM, either near the seafloor or within the ocean interior.
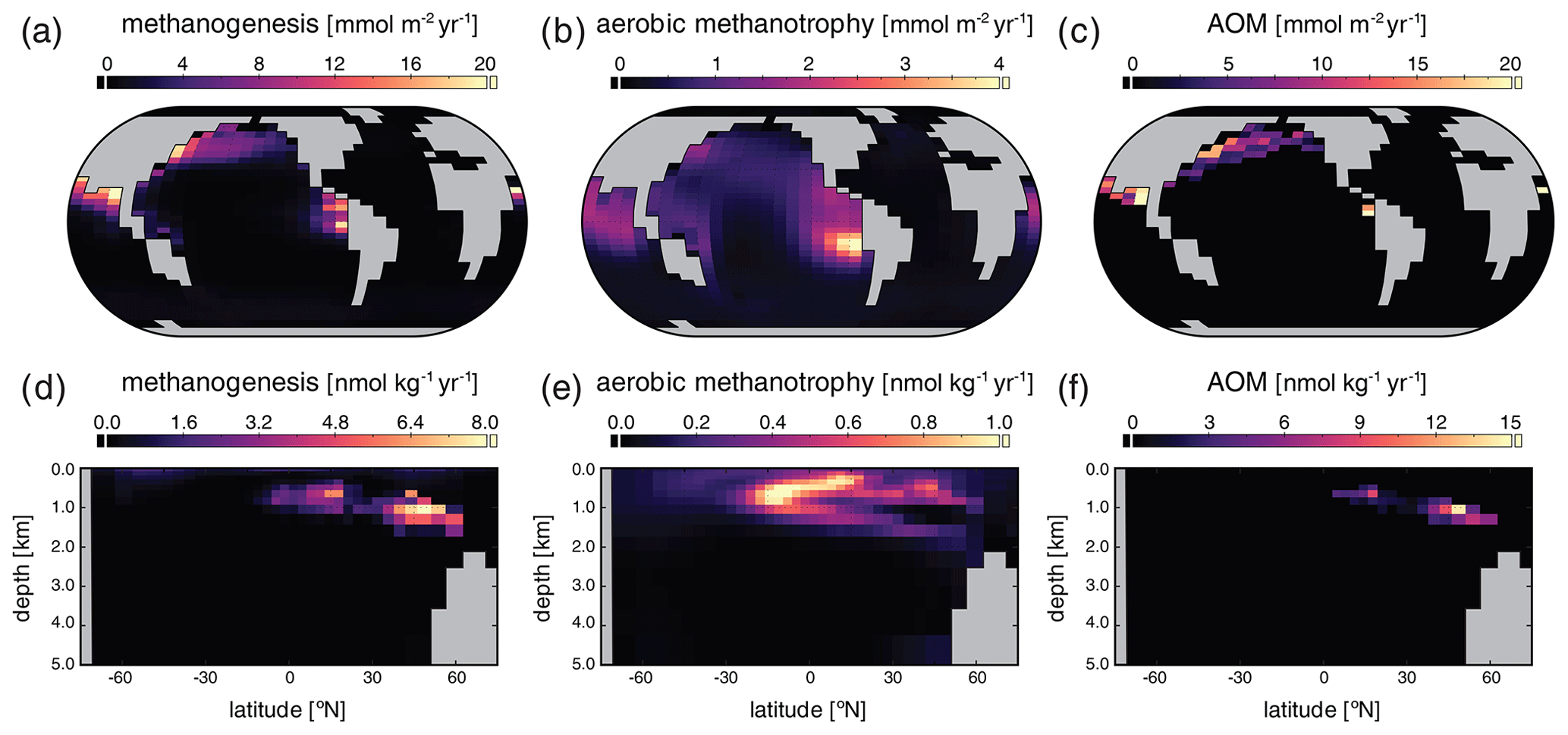
Figure 6Major biological fluxes in the marine methane cycle for our “modern” configuration. Panels show column integrated (a–c) and zonally averaged (d–f) rates of methanogenesis, aerobic methanotrophy, and anaerobic methane oxidation (AOM) in the ocean interior.
5.2 Low-pO2 (“ancient”) steady state
Next, we explore a low-pO2 steady state, similar to the Proterozoic Earth (Reinhard et al., 2017) but played out in a modern continental configuration and overturning circulation, by initializing the atmosphere with pO2, pCO2, and pCH4 of [v∕v] atm (equivalent to a value 10−3 times the present atmospheric level, PAL), 278 ppm, and 500 ppm, respectively, as well as globally uniform oceanic concentrations of SO (280 µmol kg−1) and CH4 (50 µmol kg−1). We again fix globally averaged solar insolation at the modern value (1368 W m−2), with seasonally variable forcing as a function of latitude, and set radiative forcing for CO2 and CH4 equivalent to the modern preindustrial state in order to isolate the effects of biogeochemistry on steady-state tracer distributions. The model is then spun up for 20 kyr with atmospheric pO2 and pCO2 (and δ13C of atmospheric CO2) restored to the initial values specified above at every time step, with an imposed “geologic” flux of CH4 to the atmosphere of 3 Tmol yr−1 at a δ13C value of −60 ‰. Atmospheric pCH4 and all oceanic tracers are allowed to evolve freely.
Surface, benthic, and ocean interior distributions of [O2], [SO], and [CH4] are shown in Fig. 7 for our low-pO2 simulation. Dissolved O2 concentrations are now extremely heterogeneous throughout the surface ocean, ranging over an order of magnitude from less than 1 to over 10 µmol kg−1, with concentrations that are regionally well in excess of air saturation at the prescribed pO2 of atm (Fig. 7a). Previous studies have shown that these features are not unexpected at very low atmospheric pO2 (Olson et al., 2013; Reinhard et al., 2016). We note, however, that the distribution and maximum [O2] in our low-pO2 simulation are both somewhat different from those presented in Olson et al. (2013) and Reinhard et al. (2016). We attribute this primarily to the different parameterizations of primary production in the surface ocean. In the biogeochemical configuration of cGENIE we adopt here, we allow rates of photosynthesis to scale more directly with available PO than is the case in these previous studies (Eq. 3), which allows for higher rates of oxygen production in regions of deep mixing and relatively intense organic matter recycling below the photic zone (Fig. 7a). In any case, as in previous examinations of surface [O2] dynamics at low atmospheric pO2 (Olson et al., 2013; Reinhard et al., 2016), our regional [O2] patterns still generally track the localized balance between photosynthetic O2 release and consumption through respiration and reaction with inorganic reductants, rather than temperature-dependent solubility patterns (Fig. 5a). Within the ocean interior, O2 is consumed within the upper few hundred meters and is completely absent in benthic settings (Fig. 7d, g).
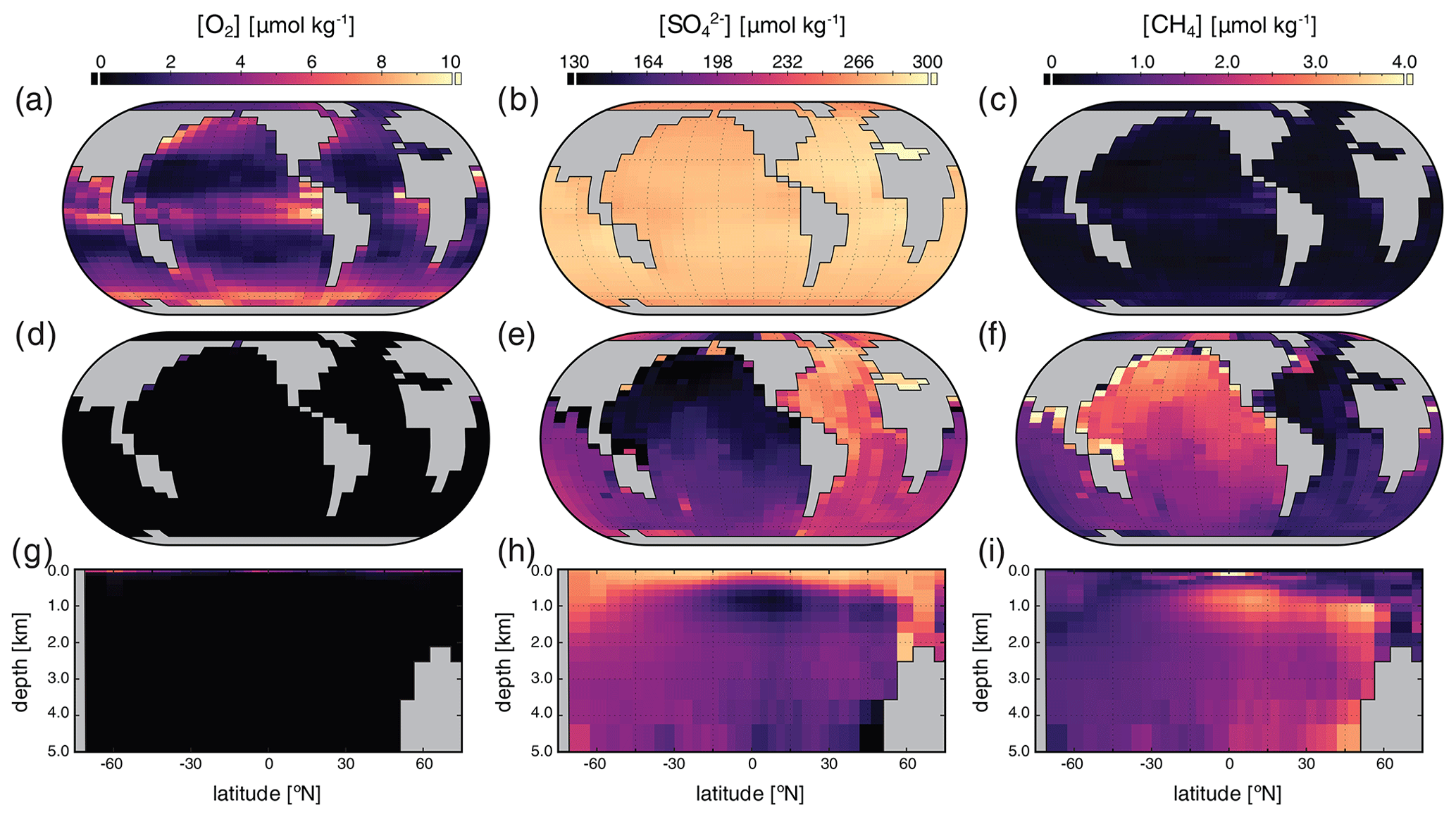
Figure 7Tracer distributions in surface (a–c) and benthic (d–f) grid cells and in the zonally averaged ocean interior (g–i) for O2 (a, d, g), SO (b, e, h), and CH4 (c, f, i) in our “ancient” configuration (see text). Note the different concentration units for each tracer and the differing scales relative to Fig. 5.
In our low-pO2 simulations we initialize the ocean with a globally uniform [SO] of 280 µmol kg−1, under the premise that the marine SO inventory should scale positively with atmospheric pO2. With this much lower initial SO inventory (i.e., 102 times less than the modern ocean), steady-state [SO] distributions are significantly more heterogeneous than in the modern, high-pO2 case (Fig. 7). Ocean [SO] is approximately homogeneous spatially in surface waters, even with a significantly reduced seawater inventory (Fig. 7a), but is strongly variable within the ocean interior (Fig. 7e, h). Indeed, in our low-pO2 simulations SO serves as the principal oxidant for organic matter remineralization in the ocean interior, with the result that its distribution effectively mirrors that of [O2] in the modern case in both spatial texture and overall magnitude (compare Fig. 7e, h with Fig. 5d, g). Dissolved SO in this simulation never drops to zero, a consequence of our initial 280 µmol kg−1 concentration of SO representing the oxidative potential of 560 µmol kg−1 of O2, some 3 times higher than the mean [O2] value in the modern ocean interior (∼ 170 µmol kg−1).
Dissolved CH4 concentrations in the surface and shallow subsurface ocean are variable but much higher than in our modern simulations, typically on the order of ∼ 1–2 µmol kg−1 (Fig. 7c). The benthic [CH4] distribution shows concentrations up to ∼ 8 µmol kg−1, with concentrations in excess of 1 µmol kg−1 pervasively distributed across the seafloor. In general, the benthic [CH4] distribution inversely mirrors that of [SO] (Fig. 7f), which results from the fact that in the low-pO2 case SO again serves as the principal oxidant of methane. Concentrations of CH4 in the ocean interior can approach ∼ 10 µmol kg−1 but in the zonal average are typically less than 5 µmol kg−1 (Fig. 7i). Overall, the oceanic CH4 inventory increases dramatically in the low-pO2 case relative to the modern simulation, from ∼ 4.5 Tmol CH4 to ∼ 1900 Tmol CH4.
The major metabolic fluxes within the ocean's microbial CH4 cycle for our “ancient” configuration are shown in Fig. 8. Column-integrated rates of microbial methanogenesis are greater than in the high-pO2 case by up to a factor of ∼ 102 (Fig. 8a), with methanogenesis also showing a much broader areal distribution. Within the ocean interior, rates of methanogenesis are most elevated in the upper ∼ 1 km (Fig. 8d) as a consequence of elevated rates of organic carbon remineralization combined with a virtual absence of dissolved O2 beneath the upper ∼ 200 m. Rates of aerobic methanotrophy, which is effectively absent in the ocean interior (Fig. 8e), are elevated relative to those observed in the high-pO2 simulation by less than an order of magnitude and are concentrated in the tropical surface ocean near the equatorial divergence (Fig. 8b). In contrast, AOM is strongly coupled spatially to microbial methanogenesis, with rates that are often well over ∼ 102 times higher than those observed in the high-pO2 case (Fig. 8c, f). Once again, AOM dominates the consumption of CH4 produced in the ocean interior and is extremely effective at reducing CH4 fluxes to the atmosphere. Despite a significant increase in overall oceanic CH4 burden relative to our high-pO2 simulation (see above and Fig. 7i), atmospheric pCH4 increases only modestly from ∼ 0.8 ppm to 6 ppm [v∕v], equivalent to an additional radiative forcing of only ∼ 2 W m−2, due to efficient microbial consumption in the upper ocean.
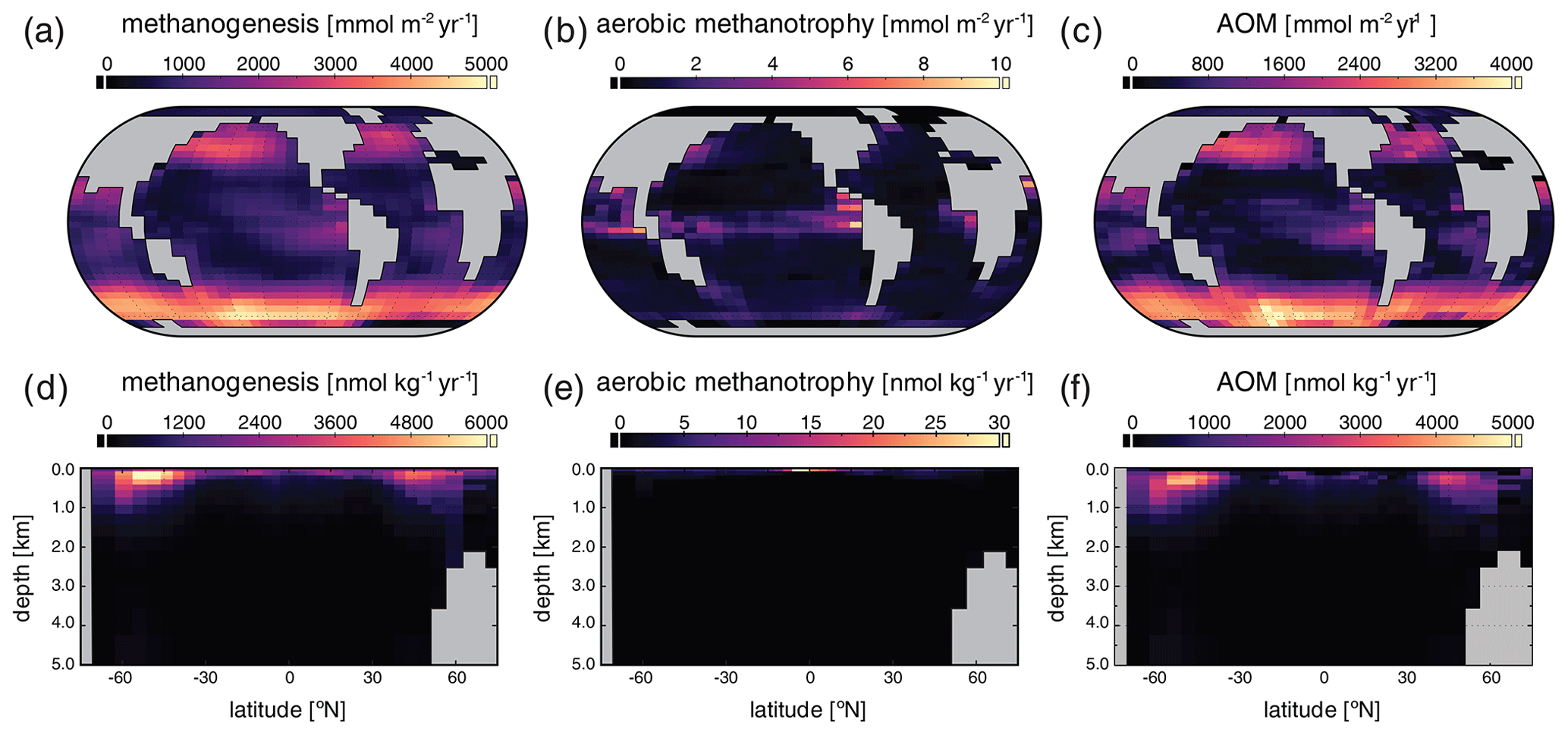
Figure 8Major biological fluxes in the marine methane cycle for our “ancient” configuration. Panels show column-integrated (a–c) and zonally averaged (d–f) rates of methanogenesis, aerobic methanotrophy, and anaerobic methane oxidation (AOM) in the ocean interior.
5.3 Atmospheric carbon injection
To illustrate the capabilities of the model in exploring the time-dependent (perturbation) behavior of the CH4 cycle, we perform a simple carbon injection experiment in which 3000 PgC is injected directly into the atmosphere either as CH4 or as CO2, starting from our modern steady state. The injection is spread over 1000 years, with an instantaneous initiation and termination of carbon input to the atmosphere. This is meant only to illustrate the time-dependent behavior of the model in the face of an idealized carbon cycle perturbation, rather than to evaluate any particular scenario for explaining previous climate transients in Earth's history. However, the magnitude and duration of this carbon injection, corresponding to 3 PgC yr−1, are meant to roughly mimic the upper end of estimates for the Paleocene–Eocene Thermal Maximum (PETM), a transient global warming event at ∼ 56 Ma hypothesized to have been driven by emissions of CO2 and/or CH4 (Kirtland Turner, 2018). This flux is much lower than the current anthropogenic carbon input of ∼ 10 PgC yr−1 (Ciais et al., 2013). For simplicity, and because we focus on only the first 3000 years following carbon injection, we treat the ocean–atmosphere system as closed, with the result that all injected carbon ultimately accumulates within the ocean and atmosphere rather than being removed through carbonate compensation and silicate weathering.
Following a carbon release to the atmosphere in the form of CH4, there is an immediate and significant increase in atmospheric pCH4 to values greater than 10 ppmv, followed by a gradual increase to a maximum of ∼ 12 ppmv throughout the duration of the CH4 input (Fig. 9a). Much of this methane is exchanged with the surface ocean and consumed by aerobic methanotrophy, while some is photochemically oxidized directly in the atmosphere, both of which lead to a significant but delayed increase in atmospheric pCO2 (Fig. 9b). This increase in atmospheric pCH4 and pCO2 leads to an increase in global average surface air temperature (SAT) of ∼ 7 ∘C (Fig. 9d) and an increase in mean ocean temperature (MOT) of ∼ 2 ∘C (Fig. 9e), along with significant acidification of the surface ocean (Fig. 9c).
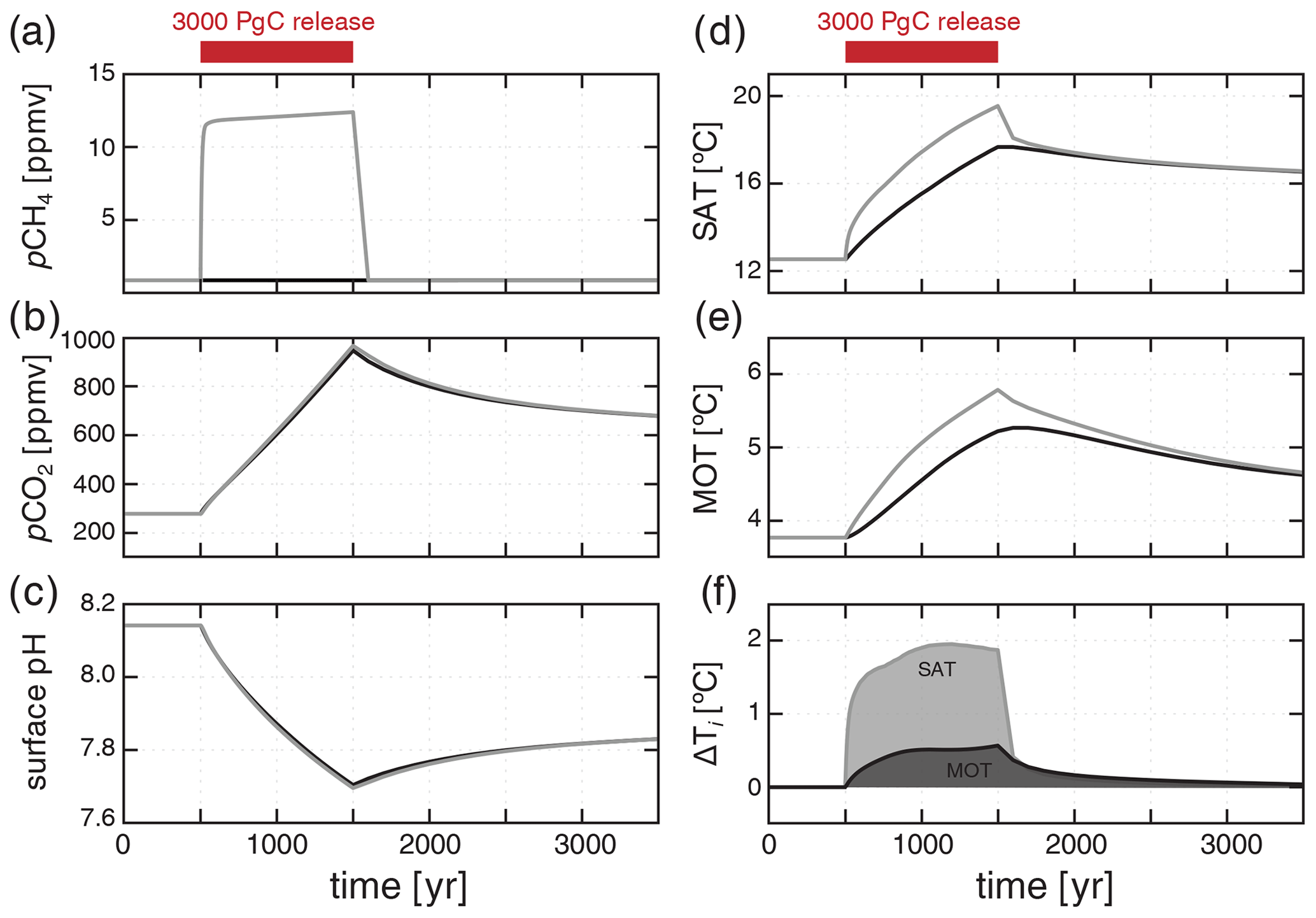
Figure 9Response to a 3000 PgC release directly to the atmosphere spread over 1000 years, assuming carbon is injected as either CH4 or CO2. Atmospheric pCH4 (a), pCO2 (b), mean surface ocean pH (c), mean surface air temperature (SAT; d), and mean ocean temperature (MOT; e) are shown for a CH4 injection (grey) and a CO2 injection (black). Panel (f) shows the difference in SAT and MOT between the CH4 and CO2 injection scenarios ( – ) through time.
The increase in atmospheric pCO2 and drop in ocean pH are nearly identical if we instead inject the carbon as CO2 rather than CH4 (Fig. 9b, c). However, when carbon is injected as CH4, there is an additional transient increase in global surface air temperature of ∼ 2 ∘C and roughly 0.5 ∘C of additional whole-ocean warming for the same carbon input and duration (Fig. 9f). This results from the fact that, mole for mole, CH4 is a much more powerful greenhouse gas than is CO2, and oxidation of CH4 to CO2 is not instantaneous during the carbon release interval. Combined, these factors result in a disequilibrium situation in which a proportion of carbon released to the atmosphere remains in the form of CH4 rather than CO2, providing an enhancement of warming, especially during the duration of carbon input. This warming enhancement should be considered in past events during which CH4 release is suspected as a key driver of warming. For instance, additional warming due to CH4 forcing may help explain the apparent discrepancy between the amount of warming reconstructed by proxy records and proposed carbon forcing during the PETM (Zeebe et al., 2009).
5.4 Atmospheric pCH4 on the early Earth
Using our low-pO2 steady state as a benchmark case (Sect. 5.2), we briefly explore the sensitivity of atmospheric pCH4 to a subset of model variables. All model ensembles are initially configured with globally homogeneous marine SO and CH4 inventories, as well as a background geologic CH4 flux of 3 Tmol yr−1, and are spun up for 20 kyr with a fixed pO2 and pCO2. We report atmospheric pCH4 from the final model year. Our purpose here is not to be exhaustive or to elucidate any particular period of Earth's history, but to demonstrate some of the major factors controlling the atmospheric abundance of CH4 on a low-oxygen Earth-like planet. We present results from individual sensitivity ensembles from our benchmark low-pO2 case over the following parameter ranges: (1) atmospheric pO2 between 10−4 and 10−1 times the present atmospheric level (PAL), equivalent to roughly and atm, respectively; (2) initial marine SO inventories corresponding to globally uniform seawater concentrations between 0 and 1000 µmol kg−1; and (3) biological energy quanta (BEQ) for anaerobic methane oxidation between 5 and 30 kJ mol−1.
Results for our low-pO2 sensitivity ensembles are shown in Fig. 10. We find a similar sensitivity of atmospheric pCH4 to atmospheric pO2 to that observed by Olson et al. (2016). In particular, atmospheric CH4 abundance initially increases as atmospheric pO2 drops below modern values to roughly 2 %–3 % PAL, after which decreasing pO2 causes pCH4 to drop. This behavior is well-known from previous 1-D photochemical model analysis and arises principally from increasing production of OH via water vapor photolysis as shielding of H2O by ozone (O3) decreases at low atmospheric pO2 (Pavlov et al., 2003; Claire et al., 2006; Goldblatt et al., 2006). However, peak atmospheric pCH4 is significantly reduced in our models relative to those of Olson et al. (2016). For example, at an “optimal” atmospheric pO2 of ∼ 2.5 % PAL, Olson et al. (2016) predict a steady-state atmospheric pCH4 of ∼ 35 ppmv, while we predict a value of ∼ 10 ppmv (Fig. 10a). This difference can be attributed to our updated O2–O3–CH4 photochemistry parameterization together with a significant upward revision in the rate constant for aerobic methanotrophy. Nevertheless, our results strongly reinforce the arguments presented in Olson et al. (2016) and, taken at face value, further marginalize the role of CH4 as a significant climate regulator at steady state during most of the Proterozoic eon (between ∼ 2.5 and 0.5 Ga).
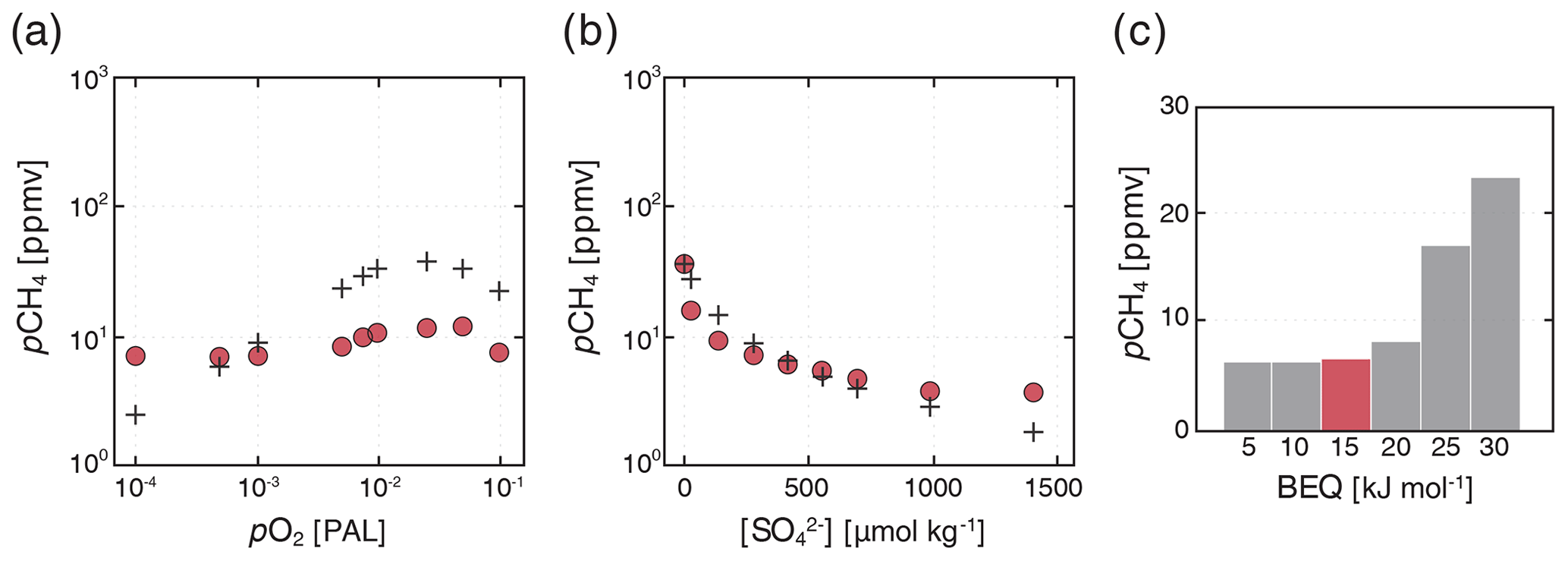
Figure 10Sensitivity ensembles of our “ancient” configuration compared to the results of Olson et al. (2016). Steady-state atmospheric pCH4 values as a function of assumed atmospheric pO2 (a) and initial marine SO inventory (b) are shown for our “ancient” configuration (filled circles; see text) and from Olson et al. (black crosses). Shown below are additional ensembles showing the impact of varying the minimum free energy yield required for microbial methane oxidation (BEQ; c) on atmospheric pCH4. All simulations were spun up from cold for 20 kyr, with the results shown from the last model year.
Atmospheric CH4 abundance is also strongly sensitive to the marine SO inventory (Fig. 10b). The scaling we observe between the initial SO inventory and steady-state atmospheric pCH4 is very similar to that reported by Olson et al. (2016), with a sharp drop in the marine CH4 inventory and atmospheric CH4 abundance as marine SO drops below ∼ 100 µmol kg−1 (Fig. 10b). The implication is that for most of Earth's history anaerobic oxidation of CH4 in the ocean interior has served as an important inhibitor of CH4 fluxes from the ocean biosphere. However, during much of the Archean eon (between 4.0 and 2.5 Ga), sulfur isotope analysis indicates that marine SO concentrations may instead have been on the order of ∼ 1–10 µmol kg−1 (Crowe et al., 2014), while atmospheric pO2 would also have been much lower than the values examined here (Pavlov and Kasting, 2002). The impact of the ocean biosphere and redox chemistry on atmospheric pCH4 and Earth's climate system may thus have been much more important prior to ∼ 2.5 billion years ago.
Atmospheric CH4 is significantly impacted by the value chosen for the biological energy quantum (BEQ). With all other parameters held constant, we observe an increase in steady-state atmospheric pCH4 from ∼ 7 to ∼ 25 ppmv when increasing the BEQ value from 20 to 30 kJ mol−1 (Fig. 10c). This effect is mediated primarily by the importance of anaerobic methanotrophy when atmospheric pO2 is low and the ocean interior is pervasively reducing. The standard free energy of AOM is of the same order of magnitude as the BEQ (see above), which elevates the importance of thermodynamic drive in controlling global rates of AOM. We would expect this effect to be much less important when aerobic methanotrophy is the predominant CH4-consuming process within the ocean biosphere, as the standard free energy of this metabolism is over an order of magnitude greater than typical BEQ values for microbial metabolism (e.g., Hoehler, 2004). In any case, our results suggest that the role of thermodynamics should be borne in mind in scenarios for which AOM is an important process in the CH4 cycle and seawater [SO] is relatively low.
The global biogeochemical cycling of CH4 is central to the climate and redox state of planetary surface environments and responds to the internal dynamics of other major biogeochemical cycles across a very wide range of spatial and temporal scales. There is thus strong impetus for the ongoing development of a spectrum of models designed to explore planetary CH4 cycling, from simple box models to more computationally expensive 3-D models with dynamic and interactive ocean circulation. Our principal goal here is the development of a mechanistically realistic but simple and flexible representation of CH4 biogeochemical cycling in Earth's ocean–atmosphere system, with the hope that this can be further developed to explore steady-state and time-dependent changes to the global CH4 cycle in Earth's past and future and ultimately to constrain CH4 cycling dynamics on Earth-like planets beyond our solar system.
To accomplish this, we have refined the organic carbon remineralization scheme in the cGENIE Earth system model to reflect the impact of anaerobic organic matter recycling in sinking aggregates within oxygenated waters and to include the carbon cycling and isotopic effects of microbial CH4 production. We have also incorporated revised schemes for microbial CH4 consumption that include both kinetic and thermodynamic constraints, and we have updated the parameterized atmospheric O2–O3–CH4 photochemistry to improve accuracy and for use across a wider range of atmospheric pO2 values than explored in previous work. Simulations of roughly modern (high-O2) and Proterozoic (low-O2) Earth system states demonstrate that the model effectively reproduces the first-order features of the modern ocean–atmosphere CH4 cycle and can be effectively implemented across a wide range of atmospheric O2 partial pressures and marine SOconcentrations. In addition, our results strongly reinforce the conclusions of Olson et al. (2016) for the Proterozoic Earth system, while going beyond this to posit that the thermodynamics of anaerobic CH4 consumption may have been important in regulating atmospheric CH4 abundance during the Archean. Finally, our simulation of PETM-like carbon injection demonstrates the importance of explicitly considering CH4 radiative forcing during transient warming events in Earth history.
We suggest that ongoing and future development work should focus on the following: (1) more rigorous tuning of organic carbon remineralization and CH4 production and consumption schemes based on data fields from the modern ocean; (2) development and implementation of a more flexible parameterization of atmospheric photochemistry that allows the roles of atmospheric temperature structure, water vapor abundance, and atmospheric pCO2 to be explored; (3) coupling of deep ocean chemistry with a description of marine methane hydrates and associated sedimentary CH4 cycling; and (4) developing a representation of the production and consumption of CH4 by terrestrial ecosystems.
A manual describing code installation, basic model configuration, and an extensive series of tutorials is provided. The Latex source of the manual and PDF file can be obtained by cloning (https://github.com/derpycode/muffindoc, last access: 15 November 2020). The user manual contains instructions for obtaining, installing, and testing the code, as well as running experiments. The version of the code used in this paper is tagged as release v0.9.14 and has the DOI https://doi.org/10.5281/zenodo.4002934 (Ridgewell et al., 2020). Configuration files for the specific experiments presented in the paper can be found in cgenie.muffin/genie-userconfigs/MS/reinhardetal.GMD.2020. Details on the different experiments, plus the command line needed to run each, are given in README.txt.
The rate data used to construct Fig. 2 are also included in the Supplement and as an accessory dataset with the DOI https://doi.org/10.5281/zenodo.4081700 (Reinhard, 2020).
The supplement related to this article is available online at: https://doi.org/10.5194/gmd-13-5687-2020-supplement.
CTR, SLO, and AR developed new model code. CTR and CP compiled and analyzed empirical data for rates of methanotrophy. CTR performed all model simulations and data analysis. CTR prepared the paper with contributions from all co-authors.
The authors declare that they have no conflict of interest.
Christopher T. Reinhard acknowledges support from the NASA Astrobiology Institute (NAI) and the NASA Nexus for Exoplanet System Science (NExSS). SLO acknowledges support from the T.C. Chamberlin Postdoctoral Fellowship in the Department of Geophysical Sciences at the University of Chicago. Andy Ridgwell, Sandra Kirtland Turner, and Yoshiki Kanzaki were supported in part by an award from the Heising–Simons Foundation. We also thank Mark Claire for providing unpublished photochemical model results.
This research has been supported by the NASA Astrobiology Institute (CAN7) and the NASA Exobiology Program (proposal 18-EXO18-0005).
This paper was edited by Fiona O'Connor and reviewed by David Archer and Jeff Ridley.
Archer, D. and Buffett, B.: Time-dependent response of the global ocean clathrate reservoir to climatic and anthropogenic forcing, Geochem. Geophys., Geosys., 6, GB1008, https://doi.org/10.1029/2004GC000854, 2005.
Archer, D., Buffett, B., and Brovkin, V.: Ocean methane hydrates as a slow tipping point in the global carbon cycle, P. Natl. Acad. Sci. USA, 106, 20596–20601, 2009.
Bartdorff, O., Wallmann, K., Latif, M., and Semenov, V.: Phanerozoic evolution of atmospheric methane, Global Biogeochem. Cy., 22, GB1008, https://doi.org/10.1029/2007GB002985, 2008.
Beerling, D., Berner, R. A., Mackenzie, F. T., Harfoot, M. B., and Pyle, J. A.: Methane and the CH4-related greenhouse effect over the past 400 million years, Am. J. Sci., 309, 97–113, 2009.
Bender, M. and Conrad, R.: Kinetics of CH4 oxidation in oxic soils exposed to ambient air or high CH4 mixing ratios, Fems. Microbiol. Lett., 101, 261–270, 1992.
Bender, M. and Conrad, R.: Kinetics of methane oxidation in oxic soils, Chemosphere, 26, 687–696, 1993.
Berner, R. A.: Activity Coefficients of Bicarbonate Carbonate and Calcium Ions in Sea Water, Geochim. Cosmochim. Ac., 29, 947–965, 1965.
Bethke, C. M., Ding, D., Jin, Q., and Sanford, R. A.: Origin of microbiological zoning in groundwater flows, Geology, 36, 739–742, 2008.
Bianchi, D., Weber, T. S., Kiko, R., and Deutsch, C.: Global niche of marine anaerobic metabolisms expanded by particle microenvironments, Nat. Geosci., 11, 263–268, 2018.
Bjerrum, C. J. and Canfield, D. E.: Towards a quantitative understanding of the late Neoproterozoic carbon cycle, P. Natl. Acad. Sci. USA, 108, 5542–5547, 2011.
Bock, M., Schmitt, J., Beck, J., Seth, B., Chappellaz, J., and Fischer, H.: Glacial/interglacial wetland, biomass burning, and geologic methane emissions constrained by dual stable isotopic CH4 ice core records, P. Natl. Acad. Sci. USA, 114, E5778–E5786, 2017.
Boetius, A., Ravenschlag, K., Schubert, C. J., Rickert, D., Widdel, F., Gieseke, A., Amann, R., Jørgensen, B. B., Witte, U., and Pfannkuche, O.: A marine microbial consortium apparently mediating anaerobic oxidation of methane, Nature, 407, 623–626, 2000.
Boudreau, B. P.: Diagenetic Models and Their Implementation, Springer-Verlag, Berlin, 1996a.
Boudreau, B. P.: A method-of-lines code for carbon and nutrient diagenesis in aquatic sediments, Comput. Geosci., 22, 479–496, 1996b.
Cao, L., Eby, M., Ridgwell, A., Caldeira, K., Archer, D., Ishida, A., Joos, F., Matsumoto, K., Mikolajewicz, U., Mouchet, A., Orr, J. C., Plattner, G.-K., Schlitzer, R., Tokos, K., Totterdell, I., Tschumi, T., Yamanaka, Y., and Yool, A.: The role of ocean transport in the uptake of anthropogenic CO2, Biogeosciences, 6, 375–390, https://doi.org/10.5194/bg-6-375-2009, 2009.
Catling, D. C., Claire, M. W., and Zahnle, K. J.: Anaerobic methanotrophy and the rise of atmospheric oxygen, Philos. T. R. Soc. A, 365, 1867–1888, 2007.
Chapelle, F. H., McMahon, P. B., Dubrovsky, N. M., Fujii, R. F., Oaksford, E. T., and Vroblesky, D. A.: Deducing the distribution of terminal electron-accepting processes in hydrologically diverse groundwater systems, Water Resour. Res., 31, 359–371, 1995.
Chronopoulou, P.-M., Shelley, F., Pritchard, W. J., Maanoja, S., and Trimmer, M.: Origin and fate of methane in the Eastern Tropical North Pacific oxygen minimum zone, ISME J., 11, 1386–1399, 2017.
Ciais, P., Sabine, C., Bala, G., Bopp, L., Brovkin, V., Canadell, J., Chhabra, A., DeFries, R., Galloway, J., Heimann, M., Jones, C., Le Quéré, C., Myneni, R. B., Piao, S., and Thornton, P.: Carbon and Other Biogeochemical Cycles, in: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, edited by: Stocker, T. F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S. K., Boschung, J., Nauels, A., Xia, Y., Bex, V., and Midgley, P. M., Cambridge University Press, Cambridge, 467–544, 2013.
Claire, M. W., Catling, D. C., and Zahnle, K. J.: Biogeochemical modelling of the rise in atmospheric oxygen, Geobiology, 4, 239–269, 2006.
Clegg, S. L. and Brimblecombe, P.: The solubility and activity coefficient of oxygen in salt solutions and brines, Geochim. Cosmochim. Ac., 54, 3315–3328, 1990.
Cramer, S. D.: Solubility of methane in brines from 0 to 300 ∘C, Ind. Eng. Chem. Proc. Dd., 23, 533–538, 1984.
Crespo-Medina, M., Meile, C. D., Hunter, K. S., Diercks, A.-R., Asper, V. L., Orphan, V. J., Tavormina, P. L., Nigro, L. M., Battles, J. J., Chanton, J. P., Shiller, A. M., Joung, D.-J., Amon, R. M. W., Bracco, A., Montoya, J. P., Villareal, T. A., Wood, A. M., and Joye, S. B.: The rise and fall of methanotrophy following a deepwater oil-well blowout, Nat. Geosci., 7, 423–427, 2014.
Crowe, S. A., Paris, G., Katsev, S., Jones, C., Kim, S., Zerkle, A. L., Nomosatryo, S., Fowle, D. A., Adkins, J. F., Sessions, A. L., Farquhar, J., and Canfield, D. E.: Sulfate was a trace constituent of Archean seawater, Science, 346, 735–739, 2014.
Curtis, G. P.: Comparison of approaches for simulating reactive solute transport involving organic degredation reactions by multiple terminal electron acceptors, Comput. Geosci., 29, 319–329, 2003.
Daines, S. J. and Lenton, T. M.: The effect of widespread early aerobic marine ecosystems on methane cycling and the Great Oxidation, Earth Planet. Sc. Lett., 434, 42–51, 2016.
Dale, A. W., Regnier, P., and Van Cappellen, P.: Bioenergetic controls on anaerobic oxidation of methane (AOM) in coastal marine sediments: A theoretical analysis, Am. J. Sci., 306, 246–294, 2006.
Dale, A. W., Regnier, P., Knab, N. J., Jørgensen, B. B., and Van Cappellen, P.: Anaerobic oxidation of methane (AOM) in marine sediments from the Skagerrak (Denmark): II. Reaction-transport modeling, Geochim. Cosmochim. Ac., 72, 2880–2894, 2008.
Dickens, G. R., Castillo, M. M., and Walker, J. C. G.: A blast of gas in the latest Paleocene: simulating first-order effects of massive dissociation of oceanic methane hydrate, Geology, 25, 259–262, 1997.
Dickens, G. R.: Rethinking the global carbon cycle with a large, dynamic and microbially mediated gas hydrate capacitor, Earth Planet. Sc. Lett., 213, 169–183, 2003.
Doney, S. C., Lindsay, K., Fung, I., and John, J.: Natural variability in a stable, 1000-yr global coupled climate-carbon cycle simulation, J. Climate, 19, 3033–3054, 2006.
Duan, Z., Møller, N., Greenberg, J., and Weare, J. H.: The prediction of methane solubility in natural waters to high ionic strength from 0 to 250∘C and from 0 to 1600 bar, Geochim. Cosmochim. Ac., 56, 1451–1460, 1992.
Dunfield, P. F. and Conrad, R.: Starvation alters the apparent half-saturation constant for methane in the Type II methanotroph Methylocystis strain LR1, Appl. Environ. Microb., 66, 4136–4138, 2000.
Edwards, N. R. and Marsh, R.: Uncertainties due to transport-parameter sensitivity in an efficient 3-D ocean-climate model, Clim. Dynam., 24, 415–433, 2005.
Egger, M., Rasigraf, O., Sapart, C. J., Jilbert, T., Jetten, M. S. M., Röckmann, T., van der Veen, C., Banda, N., Kartal, B., Ettwig, K. F., and Slomp, C. P.: Iron-mediated anaerobic oxidation of methane in brackish coastal sediments, Environ. Sci. Technol., 49, 277–283, 2015.
Egger, M., Riedinger, N., Mogollón, J. M., and Jørgensen, B. B.: Global diffusive fluxes of methane in marine sediments, Nat. Geosci., 11, 421–425, 2018.
Elliot, S., Maltrud, M., Reagan, M., Moridis, G., and Cameron-Smith, P.: Marine methane cycle simulations for the period of early global warming, J. Geophys. Res.-Biogeo., 116, G01010, https://doi.org/10.1029/2010JG001300, 2011.
Froelich, P. N., Klinkhammer, G. P., Bender, M. L., Luedtke, N. A., Heath, G. R., Cullen, D., and Duaphin, P.: Early oxidation of organic matter in pelagic sediments of the eastern equatorial Atlantic: suboxic diagenesis, Geochim. Cosmochim. Ac., 43, 1075–1090, 1979.
Goldblatt, C., Lenton, T. M., and Watson, A. J.: Bistability of atmospheric oxygen and the Great Oxidation, Nature, 443, 683–686, 2006.
Griffies, S. M.: The Gent-McWilliams skew flux, J. Phys. Oceanogr., 28, 831–841, 1998.
Hanson, R. S. and Hanson, T. E.: Methanotrophic bacteria, Microbiol. Rev., 60, 439–471, 1996.
Haqq-Misra, J., Domagal-Goldman, S. D., Kasting, P. J., and Kasting, J. F.: A revised, hazy methane greenhouse for the Archean Earth, Astrobiol., 8, 1127–1137, 2008.
Haroon, M. F., Hu, S., Shi, Y., Imelfort, M., Keller, J., Hugenholtz, P., Yuan, Z., and Tyson, G. W.: Anaerobic oxidation of methane coupled to nitrate reduction in a novel archaeal lineage, Nature, 500, 567–570, 2013.
Hayes, J. M.: Global methanotrophy at the Archean-Proterozoic transition, in: Nobel Symp. 84, Early Life on Earth, edited by: Bengston, S., Columbia University Press, New York, 220–236, 1994.
Helz, G. R., Bura-Nakíc, E., Mikac, N., and Ciglenecki, I.: New model for molybdenum behavior in euxinic waters, Chem. Geol., 284, 323–332, 2011.
Hinrichs, K.-U., Hayes, J. M., Sylva, S. P., Brewer, P. G., and DeLong, E. F.: Methane-consuming archaebacteria in marine sediments, Nature, 398, 802–805, 1999.
Hinrichs, K.-U.: Microbial fixation of methane carbon at 2.7 Ga: Was an anaerobic mechanism possible?, Geochem. Geophy. Geosy., 3, 1–10, 2002.
Hitchcock, D. R. and Lovelock, J. E.: Life detection by atmospheric analysis, Icarus, 7, 149–159, 1967.
Hoehler, T. M., Alperin, M. J., Albert, D. B., and Martens, C. S.: Field and laboratory studies of methane oxidation in an anoxic marine sediments: Evidence for a methanogen-sulfate reducer consortium, Global Biogeochem. Cy., 8, 451–463, 1994.
Hoehler, T. M., Alperin, M. J., Albert, D. B., and Martens, C. S.: Apparent minimum free energy requirements for methanogenic Archaean and sulfate-reducing bacteria in an anoxic marine sediment, Fems. Microbiol. Ecol., 38, 33–41, 2001.
Hoehler, T. M.: Biological energy requirements as quantitative boundary conditions for life in the subsurface, Geobiology, 2, 205–215, 2004.
Hunter, S. J., Goldobin, D. S., Haywood, A. M., Ridgwell, A., and Rees, J. G.: Sensitivity of the global submarine hydrate inventory to scenarios of future climate change, Earth Planet. Sc. Lett., 367, 105–115, 2013.
Jakobsen, R. and Postma, D.: Redox zoning, rates of sulfate reduction and interactions with Fe-reduction and methanogenesis in a shallow sandy aquifer, Rømø, Denmark, Geochim. Cosmochim. Ac., 63, 137–151, 1999.
Jayakumar, D. A., Naqvi, S. W. A., Narvekar, P. V., and George, M. D.: Methane in coastal and offshore waters of the Arabian Sea, Mar. Chem., 74, 1–13, 2001.
Jin, Q. and Bethke, C. M.: Predicting the rate of microbial respiration in geochemical environments, Geochim. Cosmochim. Ac., 69, 1133–1143, 2005.
Jin, Q. and Bethke, C. M.: The thermodynamics and kinetics of microbial metabolism, Am. J. Sci., 307, 643–677, 2007.
Johnson, K. S.: Carbon dioxide hydration and dehydration kinetics in seawater, Limnol. Oceanogr., 27, 849–855, 1982.
Kasting, J. F., Zahnle, K. J., and Walker, J. C. G.: Photochemistry of methane in the Earth's early atmosphere, Precambrian Res., 20, 121–148, 1983.
Kasting, J. F., Pavlov, A. A., and Siefert, J. L.: A coupled ecosystem-climate model for predicting the methane concentration in the Archean atmosphere, Origin of life and evolution of the Biosphere, 31, 271–285, 2001.
Kessler, J. D., Valentine, D. L., Redmond, M. C., Du, M., Chan, E. W., Mendes, S. D., Quiroz, E. W., Villanueva, C. J., Shusta, S. S., Werra, L. M., Yvon-Lewis, S. A., and Weber, T. C.: A persistent oxygen anomaly reveals the fate of spilled methane in the deep Gulf of Mexico, Science, 331, 312–315, 2011.
Kirschke, S., Bousquet, P., Ciais, P., Saunois, M., Canadell, J. G., Dlugokencky, E. J., Bergamaschi, P., Bergmann, D., Blake, D. R., Bruhwiler, L., Cameron-Smith, P., Castaldi, S., Chevallier, F., Feng, L., Fraser, A., Heimann, M., Hodson, E. L., Houweling, S., Josse, B., Fraser, P. J., Krummel, P. B., Lamarque, J.-F., Langenfelds, R. L., Le Quére, C., Naik, V., O'Doherty, S., Palmer, P. I., Pison, I., Plummer, D., Poulter, B., Prinn, R. G., Rigby, M., Ringeval, B., Santini, M., Schmidt, M., Shindell, D. T., Simpson, I. J., Spahni, R., Steele, L. P., Strode, S. A., Sudo, K., Szopa, S., van der Werf, G. R., Voulgarakis, A., van Weele, M., Weiss, R. F., Williams, J. E., and Zeng, G.: Three decades of global methane sources and sinks, Nat. Geosci., 6, 813–823, 2013.
Kirtland Turner, S. and Ridgwell, A.: Development of a novel empirical framework for interpreting geological carbon isotope excursions, with implications for the rate of carbon injection across the PETM, Earth Planet. Sc. Lett., 435, 1–13, 2016.
Kirtland Turner, S.: Constraints on the onset duration of the Paleocene-Eocene Thermal Maximum, Philos. T. Roy. Soc. A, 376, 20170082, https://doi.org/10.1098/rsta.2017.0082, 2018.
Konijnendijk, T. Y. M., Weber, S. L., Tuenter, E., and van Weele, M.: Methane variations on orbital timescales: a transient modeling experiment, Clim. Past, 7, 635–648, https://doi.org/10.5194/cp-7-635-2011, 2011.
Krissansen-Totton, J., Garland, R., Irwin, P., and Catling, D. C.: Detectability of biosignatures in anoxic atmospheres with the James Webb Space Telescope: A TRAPPIST-1e cast study, Astro. J., 156, 114, https://doi.org/10.3847/1538-3881/aad564, 2018.
Kuivila, K. M., Murray, J. W., and Devol, A. H.: Methane production, sulfate reduction and competition for substrates in the sediments of Lake Washington, Geochim. Cosmochim. Ac., 53, 409–416, 1989.
Lamarque, J.-F., Kiehl, J. T., Shields, C. A., Boville, B. A., and Kinnison, D. E.: Modeling the response to changes in tropospheric methane concentration: Application to the Permian-Triassic boundary, Paleoceanography, 21, PA3006, https://doi.org/10.1029/2006PA001276, 2006.
Lovley, D. R., Dwyer, D. F., and Klug, M. J.: Kinetic analysis of competition between sulfate reducers and methanogens for hydrogen in sediments, Appl. Environ. Microb., 43, 1373–1379, 1982.
Lunt, D. J., Ridgwell, A., Sluijs, A., Zachos, J. C., Hunter, S. J., and Haywood, A.: A model for orbital pacing of methane hydrate destabilization during the Palaeogene, Nat. Geosci., 4, 775–778, 2011.
Marsh, R., Müller, S. A., Yool, A., and Edwards, N. R.: Incorporation of the C-GOLDSTEIN efficient climate model into the GENIE framework: “eb_go_gs” configurations of GENIE, Geosci. Model Dev., 4, 957–992, https://doi.org/10.5194/gmd-4-957-2011, 2011.
Martens, C. S. and Berner, R. A.: Interstitial water chemistry of anoxic Long Island Sound sediments. 1. Dissolved gases, Limnol. Oceanogr., 22, 10–25, 1977.
McGlynn, S. E., Chadwick, G. L., Kempes, C. P., and Orphan, V. J.: Single cell activity reveals direct electron transfer in methanotrophic consortia, Nature, 526, 531–535, 2015.
Melton, J. R., Wania, R., Hodson, E. L., Poulter, B., Ringeval, B., Spahni, R., Bohn, T., Avis, C. A., Beerling, D. J., Chen, G., Eliseev, A. V., Denisov, S. N., Hopcroft, P. O., Lettenmaier, D. P., Riley, W. J., Singarayer, J. S., Subin, Z. M., Tian, H., Zürcher, S., Brovkin, V., van Bodegom, P. M., Kleinen, T., Yu, Z. C., and Kaplan, J. O.: Present state of global wetland extent and wetland methane modelling: conclusions from a model inter-comparison project (WETCHIMP), Biogeosciences, 10, 753–788, https://doi.org/10.5194/bg-10-753-2013, 2013.
Meyer, K. M., Ridgwell, A., and Payne, J. L.: The influence of the biological pump on ocean chemistry: implications for long-term trends in marine redox chemistry, the global carbon cycle, and marine animal ecosystems, Geobiology, 14, 207–219, 2016.
Milucka, J., Ferdelman, T. G., Polerecky, L., Franzke, D., Wegener, G., Schmid, M., Lieberwirth, I., Wagner, M., Widdel, F., and Kuypers, M. M. M.: Zero-valent sulphur is a key intermediate in marine methane oxidation, Nature, 491, 541–546, 2012.
Myhre, G., Shindell, D., Breon, F.-M., Collins, W., Fuglestvedt, J., Huang, J., Koch, D., Lamarque, J.-F., Lee, D., Mendoza, B., Nakajima, T., Robock, A., Stephens, G., Takemura, T., and Zhang, H.: Anthropogenic and natural radiative forcing, in: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, edited by: Stocker, T. F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S. K., Boschung, J., Nauels, A., Xia, Y., Bex, V., and Midgley, P. M., Cambridge University Press, Cambridge, 659–740, 2013.
Olson, S. L., Kump, L. R., and Kasting, J. F.: Quantifying the areal extent and dissolved oxygen concentrations of Archean oxygen oases, Chem. Geol., 362, 35–43, 2013.
Olson, S. L., Reinhard, C. T., and Lyons, T. W.: Limited role for methane in the mid-Proterozoic greenhouse, P. Natl. Acad. Sci. USA, 113, 11447–11452, 2016.
Orphan, V. J., House, C. H., Hinrichs, K.-U., McKeegan, K. D., and DeLong, E. F.: Methane-consuming Archaea revealed by directly coupled isotopic and phylogenetic analysis, Science, 293, 484–487, 2001.
Ozaki, K., Tajika, E., Hong, P. K., Nakagawa, Y., and Reinhard, C. T.: Effects of primitive photosynthesis on Earth's early climate system, Nat. Geosci., 11, 55–59, 2018.
Paudel, R., Mahowald, N. M., Hess, P. G. M., Meng, L., and Riley, W. J.: Attribution of changes in global wetland methane emissions from pre-industrial to present using CLM4.5-BGC, Environ. Res. Lett., 11, 034020, https://doi.org/10.1088/1748-9326/11/3/034020, 2016.
Pavlov, A. A. and Kasting, J. F.: Mass-independent fractionation of sulfur isotopes in Archean sediments: Strong evidence for an anoxic Archean atmosphere, Astrobiology, 2, 27–41, 2002.
Pavlov, A. A., Kasting, J. F., and Brown, L. L.: Greenhouse warming by CH4 in the atmosphere of early Earth, J. Geophys. Res., 105, 11981–11990, 2000.
Pavlov, A. A., Hurtgen, M. T., Kasting, J. F., and Arthur, M. A.: Methane-rich Proterozoic atmosphere?, Geology, 31, 87–90, 2003.
Prather, M. J.: Time scales in atmospheric chemistry: Theory, GWPs for CH4 and CO, and runaway growth, Geophys. Res. Lett., 23, 2597–2600, 1996.
Rabouille, C. and Gaillard, J. F.: A coupled model representing the deep-sea organic carbon mineralization and oxygen consumption in surficial sediments, J. Geophys. Res.-Oceans., 96, 2761–2776, https://doi.org/10.1029/90jc02332, 1991.
Reeburgh, W. S.: Methane consumption in Cariaco Trench waters and sediments, Earth Planet. Sc. Lett., 28, 337–344, 1976.
Reeburgh, W. S.: Oceanic methane biogeochemistry, Chem. Rev., 107, 486–513, 2007.
Regnier, P., Dale, A. W., Arndt, S., LaRowe, D. E., Mogollón, J., and Van Cappellen, P.: Quantitative analysis of anaerobic oxidation of methane (AOM) in marine sediments: A modeling perspective, Earth-Sci. Rev., 106, 105–130, 2011.
Reinhard, C. T.: Reinhard.GMD.2020.RateData [Data set], Zenodo, https://doi.org/10.5281/zenodo.4081700, 2020.
Reinhard, C. T., Planavsky, N. J., Olson, S. L., Lyons, T. W., and Erwin, D. H.: Earth's oxygen cycle and the evolution of animal life, P. Natl. Acad. Sci. USA, 113, 8933–8938, 2016.
Reinhard, C. T., Olson, S. L., Schwieterman, E. W., and Lyons, T. W.: False negatives for remote life detection on ocean-bearing planets: Lessons from the early Earth, Astrobiology, 17, 287–297, https://doi.org/10.1089/ast.2016.1598, 2017.
Ridgwell, A.: Glacial-interglacial perturbations in the global carbon cycle, PhD thesis, University of East Anglia, Norwich, UK, 2001.
Ridgwell, A., Hargreaves, J. C., Edwards, N. R., Annan, J. D., Lenton, T. M., Marsh, R., Yool, A., and Watson, A.: Marine geochemical data assimilation in an efficient Earth System Model of global biogeochemical cycling, Biogeosciences, 4, 87–104, https://doi.org/10.5194/bg-4-87-2007, 2007.
Ridgwell, A. J., Marshall, S. J., and Gregson, K.: Consumption of atmospheric methane by soils: A process-based model, Global Biogeochem. Cy., 13, 59–70, 1999.
Sagan, C. and Mullen, G.: Earth and Mars: Evolution of atmospheres and surface temperatures, Science, 177, 52–56, 1972.
Sagan, C., Thompson, W. R., Carlson, R., Gurnett, D., and Hord, C.: A search for life on Earth from the Galileo spacecraft, Nature, 365, 715–721, 1993.
Sansone, F. J., Popp, B. N., Gasc, A., Graham, A. W., and Rust, T. M.: Highly elevated methane in the eastern tropical North Pacific and associated isotopically enriched fluxes to the atmosphere, Geophys. Res. Lett., 28, 4567–4570, 2001.
Schink, B.: Energetics of syntrophic cooperation in methanogenic degradation, Microbiol. Mol. Biol. R., 61, 262–280, 1997.
Schmidt, G. A. and Shindell, D. T.: Atmospheric composition, radiative forcing, and climate change as a consequence of a massive methane release from gas hydrates, Paleoceanography, 18, 1004, https://doi.org/10.1029/2002PA000757, 2003.
Schrag, D. P., Berner, R. A., Hoffman, P. F., and Halverson, G. P.: On the initiation of a snowball Earth, Geochem. Geophys. Geosyst., 3, https://doi.org/10.1029/2001GC000219, 2002.
Scranton, M. I. and Brewer, P. G.: Consumption of dissolved methane in the deep ocean, Limnol. Oceanogr., 23, 1207–1213, 1978.
Shindell, D. T., Pechony, O., Voulgarakis, A., Faluvegi, G., Nazarenko, L., Lamarque, J.-F., Bowman, K., Milly, G., Kovari, B., Ruedy, R., and Schmidt, G. A.: Interactive ozone and methane chemistry in GISS-E2 historical and future climate simulations, Atmos. Chem. Phys., 13, 2653–2689, https://doi.org/10.5194/acp-13-2653-2013, 2013.
Sivan, O., Adler, M., Pearson, A., Gelman, F., Bar-Or, I., John, S. G., and Eckert, W.: Geochemical evidence for iron-mediated anaerobic oxidation of methane, Limnol. Oceanogr., 56, 1536–1544, 2011.
Stoessell, R. K. and Byrne, P. A.: Salting-out of methane in single-salt solutions at 25∘C and below 800 psia, Geochim. Cosmochim. Ac., 46, 1327–1332, 1982.
Thamdrup, B., Steinsdóttir, H. G. R., Bertagnolli, A. D., Padilla, C. C., Patin, N. V., Garcia-Robledo, E., Bristow, L. A., and Stewart, F. J.: Anaerobic methane oxidation is an important sink for methane in the ocean's largest oxygen minimum zone, Limnol. Oceanogr., 64, 2569–2585, https://doi.org/10.1002/lno.11235, 2019.
Thompson, A. M. and Cicerone, R. J.: Possible perturbations to atmospheric CO, CH4, and OH, J. Geophys. Res., 91, 10853–10864, 1986.
Ueno, Y., Yamada, K., Yoshida, N., Maruyama, S., and Isozaki, Y.: Evidence from fluid inclusions for microbial methanogenesis in the early Archaean era, Nature, 440, 516–519, 2006.
Ulfsbo, A., Abbas, Z., and Turner, D. R.: Activity coefficients of a simplified seawater electrolyte at varying salinity (5–40) and temperature (0 and 25∘C) using Monte Carlo simulations, Mar. Chem., 171, 78–86, 2015.
Valentine, D. L.: Emerging topics in marine methane biogeochemistry, Annu. Rev. Mar. Sci., 3, 147–171, 2011.
van Bodegom, P., Stams, F., Liesbeth, M., Boeke, S., and Leffelaar, P.: Methane oxidation and the competition for oxygen in the rice rhizosphere, Appl. Environ. Microb., 67, 3586–3597, 2001.
Van Cappellen, P., Gaillard, J.-F., and Rabouille, C.: Biogeochemical transformations in sediments: Kinetic models of early diagenesis, in: Interactions of C, N, P and S Biogeochemical Cycles and Global Change, Springer-Verlag, Berlin, 401–445, 1993.
Walter, B. P. and Heimann, M.: A process-based, climate-sensitive model to derive methane emissions from natural wetlands: Application to five wetland sites, sensitivity to model parameters, and climate, Global Biogeochem. Cy., 14, 745–765, 2000.
Wania, R., Ross, I., and Prentice, I. C.: Implementation and evaluation of a new methane model within a dynamic global vegetation model: LPJ-WHyMe v1.3.1, Geosci. Model Dev., 3, 565–584, https://doi.org/10.5194/gmd-3-565-2010, 2010.
Weber, T., Wiseman, N. A., and Kock, A.: Global ocean methane emissions dominated by shallow coastal waters, Nat. Commun., 10, 4584, https://doi.org/10.1038/s41467-019-12541-7, 2019.
Zeebe, R. E., Zachos, J. C., and Dickens, G. R.: Carbon dioxide forcing alone insufficient to explain Palaeocene-Eocene Thermal Maximum warming, Nat. Geosci., 2, 576–580, 2009.
Zeebe, R. E.: What caused the long duration of the Paleocene-Eocene Thermal Maximum?, Paleoceanography, 28, 1–13, https://doi.org/10.1002/palo.20039, 2013.
- Abstract
- Introduction
- The GENIE–cGENIE Earth system model
- Oceanic methane cycling
- Atmospheric methane cycling
- Example applications of the new capabilities in the cGENIE model
- Discussion and conclusions
- Code availability
- Data availability
- Author contributions
- Competing interests
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement
- Abstract
- Introduction
- The GENIE–cGENIE Earth system model
- Oceanic methane cycling
- Atmospheric methane cycling
- Example applications of the new capabilities in the cGENIE model
- Discussion and conclusions
- Code availability
- Data availability
- Author contributions
- Competing interests
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement