the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 04 Jun 2020
| 04 Jun 2020
CAPRAM reduction towards an operational multiphase halogen and dimethyl sulfide chemistry treatment in the chemistry transport model COSMO-MUSCAT(5.04e)
Erik H. Hoffmann
Roland Schrödner
Andreas Tilgner
Ralf Wolke
A condensed multiphase halogen and dimethyl sulfide (DMS) chemistry mechanism for application in chemistry transport models is developed by reducing the CAPRAM DMS module 1.0 (CAPRAM-DM1.0) and the CAPRAM halogen module 3.0 (CAPRAM-HM3.0). The reduction is achieved by determining the main oxidation pathways from analysing the mass fluxes of complex multiphase chemistry simulations with the air parcel model SPACCIM (SPectral Aerosol Cloud Chemistry Interaction Model). These simulations are designed to cover both pristine and polluted marine boundary layer conditions. Overall, the reduced CAPRAM-DM1.0 contains 32 gas-phase reactions, 5 phase transfers, and 12 aqueous-phase reactions, of which two processes are described as equilibrium reactions. The reduced CAPRAM-HM3.0 contains 199 gas-phase reactions, 23 phase transfers, and 87 aqueous-phase reactions. For the aqueous-phase chemistry, 39 processes are described as chemical equilibrium reactions. A comparison of simulations using the complete CAPRAM-DM1.0 and CAPRAM-HM3.0 mechanisms against the reduced ones indicates that the relative deviations are below 5 % for important inorganic and organic air pollutants and key reactive species under pristine ocean and polluted conditions. The reduced mechanism has been implemented into the chemical transport model COSMO-MUSCAT and tested by performing 2D simulations under prescribed meteorological conditions that investigate the effect of stable (stratiform cloud) and more unstable meteorological conditions (convective clouds) on marine multiphase chemistry. The simulated maximum concentration of HCl is of the order of 109 molecules cm−3 and that of BrO is around 1×107 molecules cm−3, reproducing the range of ambient measurements. Afterwards, the oxidation pathways of DMS in a cloudy marine atmosphere have been investigated in detail. The simulations demonstrate that clouds have both a direct and an indirect photochemical effect on the multiphase processing of DMS and its oxidation products. The direct photochemical effect is related to in-cloud chemistry that leads to high dimethyl sulfoxide (DMSO) oxidation rates and a subsequently enhanced formation of methane sulfonic acid compared to aerosol chemistry. The indirect photochemical effect is characterized by cloud shading, which occurs particularly in the case of stratiform clouds. The lower photolysis rate affects the activation of Br atoms and consequently lowers the formation of BrO radicals. The corresponding DMS oxidation flux is lowered by up to 30 % under thick optical clouds. Moreover, high updraught velocities lead to a strong vertical mixing of DMS into the free troposphere predominately under cloudy conditions. The photolysis of hypohalous acids (HOX, X = Cl, Br, or I) is reduced as well, resulting in higher HOX-driven sulfite-to-sulfate oxidation in aerosol particles below stratiform clouds. Altogether, the present model simulations have demonstrated the ability of the reduced mechanism to be applied in studying marine aerosol–cloud processing effects in regional models such as COSMO-MUSCAT. The reduced mechanism can be used also by other regional models for more adequate interpretations of complex marine field measurement data.
- Article
(5607 KB) - Full-text XML
-
Supplement
(6977 KB) - BibTeX
- EndNote





In the marine and coastal atmosphere the chemical composition of the gas phase, particles, and clouds as well as the size-distribution of particles are significantly influenced by emissions of sea spray aerosols (SSA) and volatile organic compounds from the sea surface (Simpson et al., 2015; Farmer et al., 2015; Quinn et al., 2015). Sea salt is an important compound of SSA (Quinn et al., 2015) and represents a primary source for reactive chlorine and bromine compounds in the troposphere (Saiz-Lopez and von Glasow, 2012; Simpson et al., 2015). For reactive iodine compounds, however, emissions of gaseous iodine compounds from the ocean surface dominate (Carpenter et al., 2012, 2013; Carpenter and Nightingale, 2015; Saiz-Lopez et al., 2012). Additionally, the ocean is the main source for dimethyl sulfide (DMS), which is the biggest natural source for atmospheric sulfur (Andreae, 1990; Lana et al., 2011). The oxidation of DMS is the key to understanding the natural radiative forcing as it affects both aerosol and cloud condensation nuclei (CCN) concentrations (Charlson et al., 1987). The chemical systems of halogens and DMS interact with each other strongly and are highly influenced by multiphase chemistry (Barnes et al., 2006; Hoffmann et al., 2016; von Glasow and Crutzen, 2004). As oceans cover around 70 % of the Earth's surface (Joshi et al., 2017; Law et al., 2013) and are in strong interaction with densely populated coastal areas (Kummu et al., 2016; von Glasow et al., 2013), this ocean-related atmospheric chemical subsystem is important for both Earth's climate and air quality.
The chemistry of reactive halogen compounds as well as of DMS is very sensitive to anthropogenic pollution. The advection of NOx and ozone has strong effects on the activation of reactive halogen compounds (Hoffmann et al., 2019b; Shechner and Tas, 2017; Mahajan et al., 2009b, a; McFiggans et al., 2002) and on DMS oxidation (Breider et al., 2010; Barnes et al., 2006; Chen et al., 2018). Moreover, reactive halogen compounds can significantly influence the depletion of NOx, ozone, SO2, volatile organic compounds (VOCs), and oxidized volatile organic compounds (OVOCs) (von Glasow et al., 2002b, a; Sherwen et al., 2017, 2016; Schmidt et al., 2016). As the NO3 radical concentration in anthropogenically influenced atmospheric environments is enhanced (Brown and Stutz, 2012), the NO3 radical-related DMS oxidation is reinforced (Breider et al., 2010; Chen et al., 2018), which influences the formation of sulfate aerosol particles and correspondingly leads to an increase of aerosol acidity (Muniz-Unamunzaga et al., 2018). The changed aerosol acidity further affects the formation of secondary organic aerosol (SOA) (Surratt et al., 2010, 2007; Gaston et al., 2014) as well as the activation of reactive halogen compounds (Keene et al., 1998). Apart from that, the ongoing reduction of fossil fuel combustion emissions in some parts of the world will promote the oxidation of DMS as an important contributor to the formation of sulfate aerosol particles even in the Northern Hemisphere (Perraud et al., 2015). Therefore, it is important that chemical transport models (CTMs) treat the crucial multiphase chemistry pathways of both reactive halogen compounds and DMS.
Currently, only a couple of multiphase chemistry mechanisms of halogens and DMS have been developed and applied within CTMs, e.g. EMAC, CAM–MECCA, and GEOS-Chem (Chen et al., 2017, 2018; Jöckel et al., 2016; Long et al., 2014). Nevertheless, the applied model core of these CTMs does not treat aqueous-phase chemistry of halogens and DMS by default. In CTMs that deal with the chemistry in the marine boundary layer (MBL) and the free troposphere, the activation of reactive halogen compounds and its dependence on aerosol acidity is often described by heterogeneous reactions. The parameters of these reactions have been determined in laboratory studies for aerosol solutions that are more ideal than they naturally occur, e.g. pure sulfate or sodium chloride/bromide aerosol. Hence, the accuracy of the description of these processes is restricted, and it cannot easily be assumed that they are representative under heterogeneous atmospheric conditions (Ammann et al., 2013). The treatment of multiphase chemistry in models allows for more detailed investigations concerning complex sea spray aerosol matrices. However, the level of detail for the implementation of aqueous-phase chemistry into CTMs is limited because of numerical restrictions, since the implementation of aqueous-phase chemistry usually consumes huge amounts of CPU time. Consequently, mostly only specific small subsystems are investigated, including a low number of halogen or DMS multiphase chemistry reactions (Chen et al., 2018, 2017). Both discussed aspects, consumption of CPU time and investigating only small subsystems, highlight that an overall picture of multiphase marine chemistry cannot be drawn by chemical transport modelling yet and might lead to an over- or underestimation of important chemical pathways.
To achieve the goal of adequately treating the multiphase chemistry of DMS and reactive halogen compounds within CTMs, not only is a solution for the high CPU consumption necessary but also the development of a condensed multiphase chemistry mechanism dealing with the complexity of these chemical systems. Currently, such an adequate mechanism does not exist and should be derived by reducing detailed multiphase chemistry mechanisms, because important chemical pathways could otherwise be missed resulting in a misinterpretation of field data.
In the present study, a reduced multiphase chemistry mechanism describing halogen and DMS chemistry is developed through a manual reduction using box model studies with the CAPRAM halogen module 3.0 (CAPRAM-HM3.0; Hoffmann et al., 2019a) and the CAPRAM DMS module 1.0 (CAPRAM-DM1.0; Hoffmann et al., 2016). Both modules currently contain the most detailed mechanisms dealing with the multiphase chemistry of these chemical systems. During the reduction procedure, two mechanisms are derived, which are afterwards combined into a single one. The combined reduced mechanism is implemented into the CTM MUSCAT (MUltiScale Chemistry Aerosol Transport; Wolke et al., 2004, 2012), which now treats detailed marine multiphase chemistry. Finally, the combined reduced mechanism is applied in idealized 2D simulations with a focus on multiphase DMS oxidation in the MBL and the various effects of clouds on halogens and DMS.
2.1 Model setup
The reduction of the marine multiphase chemistry modules CAPRAM-DM1.0 and CAPRAM-HM3.0 is achieved through modelling studies with the air parcel model SPACCIM (SPectral Aerosol Cloud Chemistry Interaction Model; Sehili et al., 2005; Wolke et al., 2005). SPACCIM is a model framework designed to solve complex multiphase chemistry systems and has already been used for the development of reduced aqueous-phase chemistry mechanisms (Deguillaume et al., 2010). The description of the simultaneously occurring chemical and physical processes in tropospheric cloud droplets and aqueous aerosol particles in SPACCIM is realized by combining a complex size-resolved cloud microphysical model and a detailed multiphase chemistry model. The standard atmospheric multiphase chemistry is represented by the near-explicit gas-phase mechanism MCMv3.2 (Jenkin et al., 2003; Saunders et al., 2003) and the near-explicit aqueous-phase mechanism CAPRAM4.0 (Bräuer et al., 2019).
The goal of reducing CAPRAM-DM1.0 and CAPRAM-HM3.0 is that both modules can be applied in different marine atmospheric environments in CTMs. To this end, simulations are carried out under two environmental conditions: (i) pristine ocean and (ii) polluted coastal area. The simulations run for 48 h and are equivalent to former simulations of atmospheric marine environments studied with CAPRAM (Bräuer et al., 2013; Hoffmann et al., 2016, 2019b).
In the simulations with pristine ocean conditions, an air parcel is moved along a predefined trajectory at a 900 hPa pressure level. The simulations are carried out at different latitudes (15, 30, 45, 60, and 75∘) and in different seasons of the year (summer and winter). The air temperature in the simulations is adjusted accordingly. Furthermore, the simulations are performed at different relative humidity levels (50 %, 70 %, and 90 %). In the simulations with relative humidity levels of 70 % and 90 %, cloud passages of the air parcel are considered. The cloud occurrence is modelled by uplifting the air parcel to an 800 hPa pressure level either at noon and midnight or in the early morning and evening of the first model day. Due to adiabatic cooling, the relative humidity increases, reaching the critical supersaturation so that a cloud is formed. After the cloud passage, the air parcel descends again to 900 hPa. The updraught and downdraught of the air parcel requires 0.5 h of modelling time in each case. The in-cloud residential time of the air parcel is 2 h in the simulations with cloud occurrences at noon and midnight and 3 h in those with cloud occurrences in the early morning and evening of the first model day. The simulations with 50 % relative humidity have no cloud passages of the air parcel included. For a detailed description of the emission and initialization of chemical species within the pristine ocean scenario, the reader is referred to Bräuer et al. (2013) and Hoffmann et al. (2016). The modelled concentration time profiles of specific important trace gases and aerosol compounds within the pristine boundary layer are given in Figs. S1–S10 in the Supplement.
The scenario at the polluted coastal area is divided into two sub-simulations at 45∘ latitude and 70 % relative humidity. The lower diversity of the simulations compared to the pristine ocean scenario is chosen because previous model studies had revealed that high NOx concentrations suppress gas-phase halogen radical cross-reactions and lead to a domination of halogen nitrate and nitryl chloride photolysis in halogen atom activation (Hoffmann et al., 2019a, b; Faxon and Allen, 2013; Saiz-Lopez and von Glasow, 2012; von Glasow et al., 2013). The effect of photolysis and temperature change does not affect these four important halogen activation precursors (ClNO2, ClNO3, BrNO3, and INO3). In the first simulation, the air parcel represents the composition of a pristine marine environment, which is advected over a polluted coastal urban area. The second simulation describes an air–sea breeze circulation system. Details on the model configurations of the first simulation are explicitly given in Hoffmann et al. (2019b) and those of the second simulation in Hoffmann et al. (2019a). The main model results of both simulations do not differ from the present work.
The reduction of both modules is performed by analysing the modelled 10 min time-resolved source and sink fluxes of key chemical compounds of marine multiphase chemistry. These cover all DMS oxidation intermediates and, for halogen chemistry, all XY species (X, X2O2, XNO2, XNO3, XO, XOO, OXO, XY, HOX, and HX, with X and/or Y = Cl, Br, or I). At first, CAPRAM-DM1.0 is reduced and afterwards CAPRAM-HM3.0. In the following Sect. 2.2 and 2.3, the development of the reduced CAPRAM-DM1.0 and CAPRAM-HM3.0 is described.
The final reduced marine multiphase chemistry mechanism is evaluated through control simulations, which are carried out by using not only the full but also the reduced marine multiphase chemistry mechanism.
The goal of the mechanism reduction firstly is that the modelled concentration of chemical species that are classically treated as important in CTMs, e.g. ozone, SO2, NOx, sulfate, or nitrate, only deviate from the modelled concentration obtained from the complete scheme by less than 5 % on average over the full modelling time. Secondly, concentrations of oxidants and important chemical compounds of marine multiphase chemistry should only differ by less than 10 % on average. Important chemical compounds of DMS multiphase oxidation are DMS, dimethyl sulfoxide (DMSO), and methane sulfonic acid (MSA), which represent the key stable compounds from DMS oxidation. Dimethyl sulfone (DMSO2) is not considered, because, according to current scientific knowledge, the oxidation of DMSO2 is negligible under atmospheric conditions of the pristine ocean. Additionally, the deviation of the concentration of methane sulfinic acid (MSIA) is not a reduction criterion. MSIA is very reactive, so even slight changes will immediately result in differences in the MSA and sulfate concentrations. In the case of halogen multiphase chemistry, important species for the mechanism reduction are the Cl, Br, and I atoms as well as the ClO, BrO, and IO radical and stable halogen compounds, which can act as important reservoir or activation species for halogen radicals, i.e. hypohalous acids, nitryl chloride, and dihalogen molecules. These halogen reservoir or activation species are of high importance as strong changes in their budget will obviously affect the overall oxidation processes in the MBL. For other halogen radicals, it has been shown by previous studies (e.g. see Saiz-Lopez et al., 2012; Simpson et al., 2015) that these are rapidly converted into the above-mentioned compounds and thus strong concentration changes will show up in the concentration of X atoms or XO radicals. Lastly, the pattern of the concentration time profile for all species has to match between the reduced and the full mechanism (R2≥0.75).
2.2 Development of the reduced multiphase DMS chemistry module
The oxidation of DMS in the tropospheric multiphase system leads to gaseous SO2, sulfuric acid, DMSO2, MSA, dissolved sulfate, or methane sulfonate through a sequence of steps (Hoffmann et al., 2016; Barnes et al., 2006). To cover the important intermediate oxidation steps, the reduction consists of six individual ones:
-
consideration of main multiphase DMS oxidation pathways only;
-
lumping of simple reactions;
-
application of the pseudo-steady-state approximation;
-
neglect of production and oxidation of DMSO2 in the aqueous phase;
-
lumping of the aqueous-phase oxidation of MSIA;
-
reduction of oxidation and production pathways of specific chemical compounds unimportant in the gas or aqueous phase.
In the following, these mechanism reduction steps are outlined in more detail.
2.2.1 Main pathways of multiphase DMS oxidation
In the first reduction step, the main pathways of the multiphase oxidation of DMS and its oxidation products are investigated by analysing the time-resolved source and sink fluxes of all simulations. Main pathways are defined here as chemical production or loss processes that contribute more than 5 % to the overall average mass flux of the investigated compound. This analytical approach has proven its applicability in manual mechanism reduction (Deguillaume et al., 2010; Ervens et al., 2003). To provide a reduced mechanism applicable for a wide range of conditions, a chemical reaction is included in the reduced scheme in any case if the reaction is important under a single simulation condition. The present analyses provide the important DMS multiphase reaction pathways within the troposphere, similar to a former CAPRAM study dealing with multiphase DMS chemistry (Hoffmann et al., 2016). This approach determines the subsequent reduction steps.
2.2.2 Lumping of simple reactions
According to current knowledge, in the gas phase, the oxidation of DMS or its oxidation products through H-atom abstraction leads to a corresponding peroxyl radical, which can be further oxidized into an alkoxy radical. Recently, it was suggested that the methylthiomethyl peroxyl radical (CH3SCH2O2) undergoes a rapid unimolecular H-shift (Wu et al., 2015; Berndt et al., 2019). The final stable product will be an oxidized organic sulfur compound that is characterized by an aldehyde and an organic hydrogen peroxide functionality. This compound can be oxidized in both gas and aqueous phases. Currently, the chemistry of this compound is not further investigated and therefore not treated in CAPRAM-DM1.0. However, when more laboratory data are available, further mechanisms have to consider this chemistry to improve the modelling of DMS chemistry and its effects.
Then, the DMS oxidation-derived alkoxy radical can undergo thermal decomposition, a reaction with oxygen, or isomerization (Lightfoot et al., 1992). Nevertheless, the current gas-phase chemistry of CAPRAM-DM1.0 is based on MCMv3.2, which only treats thermal decomposition for the CH3SCH2O, i.e. C−C bond scission leading to the release of formaldehyde (HCHO). Since the analyses of the mass fluxes reveal that decomposition appears immediately after formation, further decomposition products are directly incorporated in every reaction in which an alkoxy radical is formed and the corresponding decomposition reaction is deleted (see Reactions R1–R3).
Further simple integrated reactions correspond to reactions of DMS oxidation products in the aqueous phase, i.e. reactions with water. The reduction of CAPRAM3.0 has already revealed that peroxyl radical formations in the aqueous phase are not reaction-rate determining steps (Deguillaume et al., 2010). The same is true if reactions of oxidation intermediates occur with water only. Therefore, such aqueous-phase reactions are deleted and the products are directly implemented on the right-hand side of the affected reaction equations.
2.2.3 Application of the pseudo-steady-state approximation
The oxidation of DMS by the OH radical and the Cl atom occurs not only through H-atom abstraction but also through the addition of these radicals onto the sulfur atom. The formed DMS adduct is unstable and decomposes back into DMS and the corresponding radical if it is not stabilized through a reaction with oxygen, which adds to the sulfur atom (Barnes et al., 2006). It is possible that the DMS adduct reacts with NOx or decomposes into methane sulfenic acid (CH3SOH) and a methyl (CH3) radical (Barnes et al., 2006; Yin et al., 1990). The first reduction step has already revealed that even at polluted coastlines, with NOx concentrations above 10 ppb, NOx-related decompositions are not of atmospheric importance though. Overall, the analysis shows that oxygen is too reactive against DMS adducts.
The pseudo-steady-state approximation (PSSA) is a fundamental way to deal with such reactive intermediates to derive the overall rate of a chemical reaction (Seinfeld and Pandis, 2006).
As outlined, apart from the DMSO formation, a considerable amount of CH3SOH is also formed through DMS adduct decomposition. Hence, a PSSA is also effective for this reaction sequence. The implemented rate constant of the DMS adduct decomposition within CAPRAM-DM1.0 is given temperature independent. The same is true for the oxygen addition reaction. Therefore, under tropospheric conditions both reactions can be aggregated into first-order reaction rate constants and merged. This merging gives a ratio between decomposition and oxygen addition that is implemented into the overall reaction (see Reactions R4–R7).
The PSSA is also applicable to the oxidation of DMSO by the Cl atom and for the methylthiyl radical (CH3S) reaction with oxygen, leading to the methylthio peroxyl radical (CH3SOO), which has multiple reaction pathways. On the one hand, CH3SOO can react with NOx or decompose back into CH3S or decompose into SO2 and CH3; on the other hand, it can rearrange itself into the sulfonyl radical (CH3SO2). The first reduction step reveals that the reaction with NOx is negligible since only a decomposition into SO2 and the formation of the sulfonyl radical occur. For that reason, the PSSA is also applied to these two reaction pathways as well.
2.2.4 Neglect of production and oxidation of DMSO2
Though the analysis of the sink and source fluxes has revealed that aqueous-phase chemistry contributes a little more than 5 % to DMSO2 formation and oxidation, the modelled overall DMSO2 formation and oxidation flux is negligible compared to that of MSIA. Furthermore, DMSO2 has low reactivity towards OH oxidation in the gas ( cm3 molecules−1 s−1; Falbe-Hansen et al., 2000) and aqueous phases ( l mol−1 s−1; Zhu et al., 2003) Because of the low measured background gas-phase concentrations of DMSO2 in the single-digit parts per trillion (ppt) range (Davis et al., 1998; Berresheim et al., 1998), the gas-phase oxidation of DMSO2 by the OH radical is likely to be suppressed through methane oxidation ( to cm3 molecules−1 s−1 in the temperature range of 270 to 298 K) and dry and wet deposition can be assumed as the major atmospheric removal processes for DMSO2. In order to shrink the mechanism, DMSO2 production in the aqueous phase as well as the oxidation of DMSO2 in both the gas and aqueous phases are neglected as a consequence.
2.2.5 Lumping of the aqueous-phase oxidation of MSIA
In the fifth reduction step, the oxidation of MSIA in the aqueous phase has been simplified. Having a pKa value of 2.28 (Wudl et al., 1967), MSIA occurs in both its non-dissociated and its dissociated form under atmospheric aerosol as well as cloud conditions. At this point, the only important oxidant for the non-dissociated form is ozone. The deprotonated MSIA reacts with both the OH and the radical via an electron transfer reaction into aqueous CH3SO2. The formed CH3SO2 further reacts with O2 into the methylsulfonylperoxyl radical (CH3SO2O2) or decomposes into the CH3 radical and dissolved SO2 that is immediately dissociated into . Because of its high atmospheric abundance, in our study the O2 concentration is modelled to be almost constant within the tropospheric aqueous phase. Furthermore, both reaction rate constants are implemented as temperature independent, which is why a ratio between these reactions can be calculated. The reaction of the CH3SO2O2 with MSIA yields MSA and the methylsulfonylalkoxyl radical (CH3SO3). The latter decomposes into the CH3 radical and sulfate. Both reactions occur immediately. As a whole, all reactions of the deprotonated MSIA oxidation can be summarized into one reaction for each oxidant, covering the overall MSIA loss (see below Reactions R8–R13 for MSIA oxidation by the OH radical).
2.2.6 Reduction of oxidation and production pathways of specific chemical compounds less important in gas or aqueous phase
In the last reduction step, the mechanism is again analysed for residual multiphase chemistry pathways to be combined. These are, for example, reactions of radicals that now treat only one fast reaction sequence and thus are merged into the previous reaction.
2.3 Development of the reduced multiphase halogen chemistry module
The goal of reducing CAPRAM-HM3.0 is to enable the description of key halogen chemistry affecting ozone, NOx, SO2, VOCs, and OVOCs by a reduced mechanism that almost conserves the concentration time profile of the reactive halogen compounds listed earlier. The following three reduction steps are applied to achieve this:
-
consideration of chemical production or loss processes that contribute more than 5 % to the overall mass flux of halogen compounds only;
-
lumping of simple reaction sequences;
-
neglect of oxidation and production of specific chemical species modelled to be unimportant in tropospheric gas- or aqueous-phase chemistry.
In the following, these mechanism reduction steps are outlined in more detail.
2.3.1 Main pathways of multiphase halogen chemistry
An analysis of the main pathways is performed in a similar manner as for the multiphase DMS chemistry. However, the development of a DMS-like stepwise oxidation scheme is impossible, due to important side pathways and interconnections with other chemical subsystems, e.g. NOx or HOx chemistry and various halogen cross-interactions (Saiz-Lopez and von Glasow, 2012). Furthermore, halogen multiphase chemistry is characterized by large differences in aqueous-phase oxidation within aerosol particles and cloud droplets (Bräuer et al., 2013; Hoffmann et al., 2019b; von Glasow et al., 2002a). Therefore, an appropriate reduced representation of multiphase halogen chemistry requires the focus of the reduction to be on different halogen species. Hence, the determination of the main pathways is done for a huge number of halogen compounds covering key halogen atoms, halogen radicals, halogen nitrates, halogenated organics such as halogenated aldehydes, as well as halogen oxo-carboxylic acids. Again, as for the CAPRAM-DM1.0 reduction, a chemical reaction is included in the reduced scheme in any case if the reaction is important under a single simulation condition.
As already modelled in other studies, the analyses revealed that the Cl atom is an important oxidant for VOCs and OVOCs, e.g. alkanes, non-oxidized aromatic compounds, alcohols, and aldehydes (e.g. Hoffmann et al., 2019a; Sherwen et al., 2016; Xue et al., 2015; Pechtl and von Glasow, 2007). In order to restrict computational costs, chemical mechanisms in state-of-the-art CTM applications do not contain a high number of organic compounds as the near-explicit MCM. In order to still represent the chemistry of important VOCs and OVOCs in CTMs, species of the same compound classes or of equal reactivity are typically merged into “lumped” species in condensed mechanisms applied in CTMs (Baklanov et al., 2014). Based on these limitations, the reduced CAPRAM-HM3.0 has to be linkable with the chemical mechanisms used in CTMs. A first screening on treated VOCs and OVOCs in the gas-phase chemical mechanisms MOZART4.0 (Schultz et al., 2018), RACM2 (Goliff et al., 2013), MECCA (Jöckel et al., 2016), GEOS-Chem (Wang et al., 2019), and SAPRC11 (Yan et al., 2019) has been performed for the main VOCs and OVOCs for this purpose. It has been shown that most of the mechanisms contain the same set of primary VOC∕OVOC compound classes; for example, aldehydes and alcohols are often treated up to a carbon number of two. As outlined in Sect. 3, the gas-phase mechanism MOZART4 is chosen for further modelling with COSMO-MUSCAT. As a result, only the Cl atom oxidation of the lumped VOCs and OVOCs that are treated within MOZART4 are considered further. These sets can be applied to the other mechanisms, but they have to be adjusted if species are missing. The chosen reaction rate constant is based on the first lumped product. As the k values are higher with longer carbon chain, this approach is suitable, as it gives a lower limit of the reaction rate constant. Thus, no overestimation will occur. However, when the simulations with the reduced version of CAPRAM-HM3.0 are compared to the simulations with the non-condensed CAPRAM-HM3.0, this approach results in lower HCl but higher ClO formation.
2.3.2 Lumping of simple reaction sequences
In the gas phase, halogen atoms react rapidly with O2 and CO, yielding unstable molecules that, as the model simulations show, immediately decompose again. Still, within CAPRAM-HM3.0, specific oxidation pathways lead to unstable molecules (e.g. the oxidation of halogenated oxidized organics). Consequently, in every reaction in which such an unstable molecule occurs as a product it is replaced by the halogen atom and O2 or CO.
The further processing of halogenated organic peroxyl radicals in the gas phase results in halogenated organic alkoxy radicals. As for DMS, the halogenated organic alkoxy radical decomposition, which is modelled to not be the overall rate-determining step is integrated into these reactions. Overall, the recombination of the halogenated organic peroxyl radicals with other organic peroxyl radicals (RO2) leads exclusively to halogenated carbonyls (see Reactions R14–R17).
If the analysis of the main pathways leads to only one further reaction of a compound being left, this reaction was screened for two criteria: (i) does the follow-up reaction occur rapidly and (ii) is the overall concentration of the product so low that it would not be a significant interfering factor for the modelling? If both criteria are true, the overall reaction is merged together. For example, the recombination of IO in the aqueous phase leads to iodite (HIO2), which is an intermediate in the conversion between iodide and iodate (). It is quickly oxidized into iodate by H2O2, which is ubiquitous in the marine atmospheric multiphase (Jacob and Klockow, 1992; Benedict et al., 2012; Kim et al., 2007; Yuan and Shiller, 2000), and it also has a very low modelled concentration. Overall, the IO recombination together with the oxidation of HIO2 by H2O2 results in iodate (see Reactions R18–R20).
2.3.3 Neglect of oxidation and production of specific chemical species modelled to be less important in tropospheric gas or aqueous-phase chemistry
For the reduction of the halogen chemistry part, less important chemical halogen species are defined as such with low (<0.1 ppt for non-radical species) modelled concentrations or high chemical stability ( cm3 molecules−1 s−1 that is the k298 of methane; Atkinson et al., 2006) in order that their non-consideration does not affect the concentrations of the target species under conditions in the lower troposphere. While typical species with a rather high chemical stability are chlorinated and brominated organics (e.g. CH3Cl), except bromoform (CHBr3), for which oxidation in the lower troposphere is negligible, typical species with low modelled concentrations are oxidized halogenated organics derived from the OH oxidation of methylated halogens (e.g. ICHO or ICIO from CH3I oxidation). As the reduced mechanism is developed to deal with tropospheric multiphase chemistry, the oxidation of such species is not treated within the reduced CAPRAM-HM3.0.
2.4 Evaluation of the reduction steps
By comparing simulations with the reduced and with the original CAPRAM-DM1.0 and CAPRAM-HM3.0 versions added to the multiphase chemistry mechanism MCMv3.2/CAPRAM4.0, the performed reduction steps are evaluated. Being referred to as pristine, breeze, and outflow scenarios, the evaluation simulations are carried out for 45∘ latitude with a relative humidity of 70 % under pristine ocean (Hoffmann et al., 2016) and polluted coastal conditions (Hoffmann et al., 2019a). While the scenarios pristine and breeze stand for simulations of the pristine ocean as well as of an air–sea breeze circulation system, the outflow scenario represents the advection of polluted air masses over a marine environment. The evaluation simulations run for 96 h in total including cloud passages between 11:00 and 13:00 LT (local time) and between 23:00 and 01:00 LT in both pristine and outflow scenarios as well as between 13:00 and 14:00 LT in the third breeze scenario. The longer simulation time compared to that of previous simulations was chosen in order to investigate the effect of a longer modelling time on concentration divergence. The evaluation by these three simulation cases is appropriate, because both reduced mechanisms contain reactions that were both important and not important under the different performed simulations. Thus, other possible evaluation simulations would also treat reactions that are not necessary under specific conditions. Regardless of the simulation setup, a similar performance is expected as a consequence.
The investigation of the modelled evolution of the concentration time profile of ozone is shown in Fig. 1, revealing an excellent agreement for all the scenarios (R2=1). Moreover, the average ozone concentrations diverge by less than 5 % throughout the entire modelling time, demonstrating that the reduced mechanism is able to reproduce the modelled ozone concentrations of the complex mechanism.

Figure 1Modelled concentrations of ozone within the pristine, breeze, and outflow scenarios compared between the simulations with the full (solid lines) and reduced (dotted lines) CAPRAM-DM1.0 and CAPRAM-HM3.0 mechanisms.
The same analysis is performed for other air pollutants and key aerosol compounds important for air quality modelling, which are NO, NO2, SO2, HNO3, HCl, and DMS in the gas phase, and also for the dry mass as well as the organic mass of the aerosols together with nitrate, sulfate, chloride, and methane sulfonate. Furthermore, the analysis is performed for reactive halogen compounds and the OH, NO3, and HO2 radicals. Table 1 shows the average percentage deviation for these chemical species. The main target species, except MSA in the breeze scenario, do not exceed the 5 % threshold, which is also true for the OH, HO2, and NO3 radicals in both the gas and aqueous phases. Even for reactive halogen compounds, the deviation rarely exceeds the 5 % mark.
Table 1Average percentage deviations (%) of some inorganic and organic target compounds between the simulations with the full and reduced CAPRAM-DM1.0 and CAPRAM-HM3.0 mechanisms (deviations calculated throughout the full SPACCIM simulation). Exceedances of the threshold are marked in italic.

However, especially in the outflow scenario, reactive bromine compounds exceed the 10 % threshold being caused by missing brominated organics in the reduced CAPRAM-HM3.0 that trap the bromine from further reaction. For example, 3 ppt of bromine is trapped on average in brominated alcohols formed through Br atom-related oxidation of alkenes and further RO2 recombination. Regarding the low concentration and the fact that alcohols are further oxidized into carbonyls, only the formation of brominated carbonyls is considered in the reduced CAPRAM-HM3.0 to minimize the mechanism. Consequently, the bromine radical is recycled faster by the following reaction sequence.
Overall, this increases the modelled concentrations of reactive bromine compounds, particularly in the afternoon after cloud occurrence and under high alkene as well as low ozone. However, the evolution of the concentration time profile fits very well (R2=0.98, 0.95, 0.97, and 0.75 for Br, BrO, HOBr, and Br2, respectively). Apart from bromine, larger differences also occur for HOCl in the breeze and outflow scenarios. This is related to the restricted VOC and OVOC oxidations within the reduced CAPRAM-HM3.0 in order to match the condensed gas-phase chemistry mechanisms implemented in the CTMs. Therefore, the HCl concentration is more than 2 % smaller, and more Cl atoms react with ozone to form ClO that quickly reacts with HO2 to yield HOCl. As opposed to Br2 formation by HOBr, the higher HOCl does not necessarily lead to a higher modelled Cl2 formation, which is related to the significant higher chloride content in sea spray aerosols compared to bromine. It so concludes that the enhanced HOCl seems not to be a driving factor for Cl2 formation under pristine ocean conditions.
Finally, the methane sulfonate anion (MS−) is around one-quarter lower in the breeze scenario, which is caused by the radical oxidation. This reduction revealed that the radical is an important oxidant for oxalic acid and MS−. Consequently, other OVOC oxidations are discarded for treatment in the reduced CAPRAM-HM3.0. The modelled radical concentrations are around 1 order of magnitude higher in the breeze scenario compared to the pristine and outflow scenarios, resulting in much higher MS− oxidation rates. Yet, the development of the MS concentration–time profile agrees very well (R2=1). In addition, the MSA concentration deviates by only 6 % at the end of the model simulations.
Since, the evaluation reveals that the reduced mechanism system is able to reproduce similar results as the full mechanism system, it is basically appropriate for implementation into CTMs. The reduced mechanisms of CAPRAM-DM1.0 and CAPRAM-HM3.0 will be called CAPRAM-DM1.0red and CAPRAM-HM3.0red in the following text.
Also, the modelling studies reveal that computational (CPU) time is decreased, especially within the pristine scenario (see Fig. 2). Compared to the base runs, the CPU time is reduced by 16 %, 5 %, and 6 % in the pristine, breeze, and outflow scenarios, respectively. Overall, the CPU time reduction is low, but it is because of the usage of MCMv3.2 and CAPRAM4.0 that the simulations still treat more than 21 000 reactions. Furthermore, the calculation of microphysical processes consumes a huge amount of CPU time, too. Therefore, the still high CPU time is caused by requirements of the standard multiphase chemistry mechanism. These high requirements cover the reduction of CPU time achieved by the reduction efforts. Very low initialized NOx concentrations in the pristine scenario induced the stronger decrease of computation time unlike the other scenarios. When it comes to the breeze and outflow scenarios, the high initialized NOx concentrations effectively suppress halogen radical cross-interactions in the gas phase. These cross-interactions are very fast and establish reaction cycles that induces high fluxes. Therefore, under high NOx conditions, a much lower CPU time is required to solve these reactions. However, in the pristine scenario, the rapidly occurring gas-phase cross-interactions of halogen radicals still exist, hindering stronger amplified CPU time reductions.
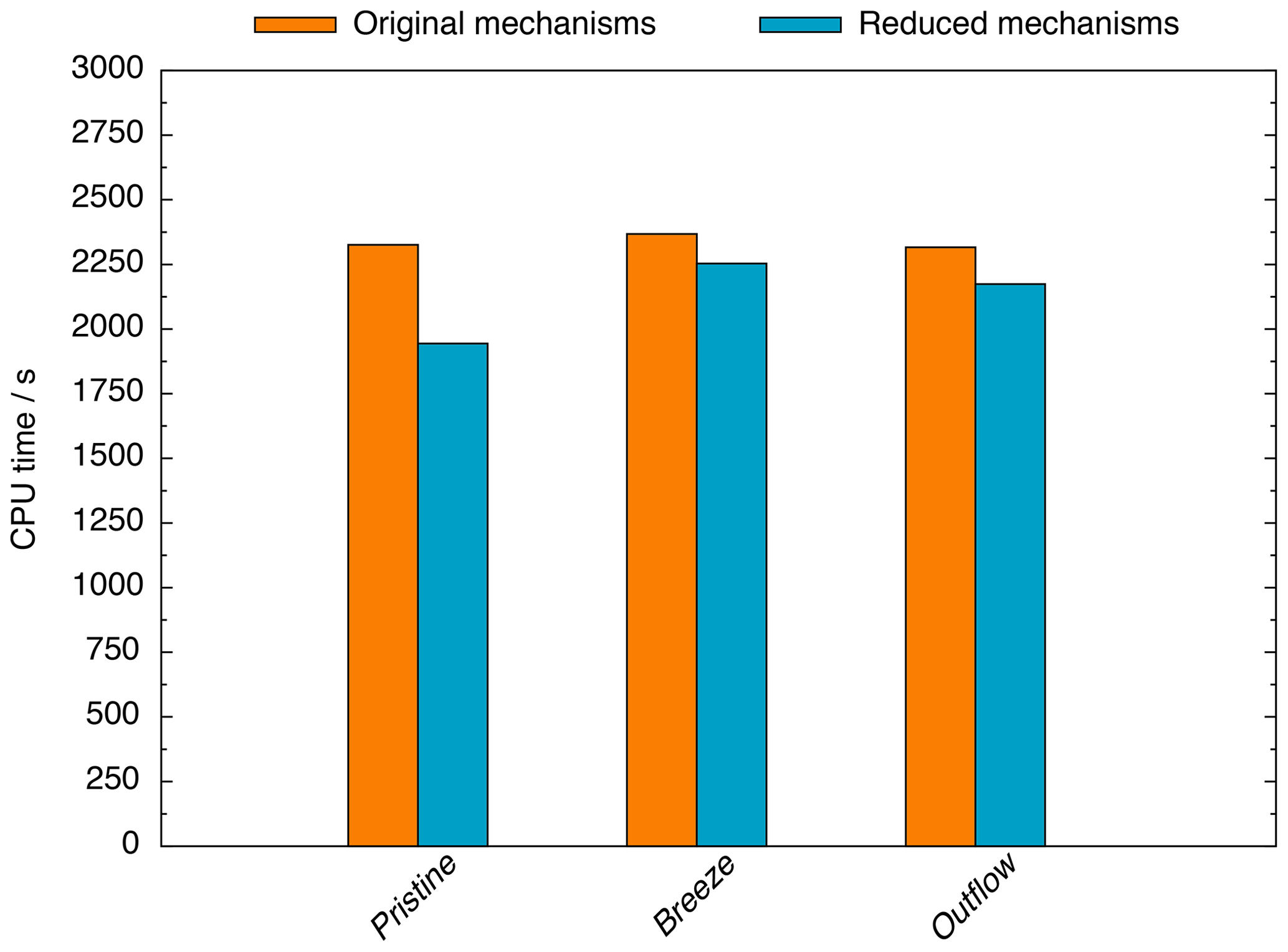
Figure 2Required CPU time within the pristine, breeze, and outflow scenarios considering the original multiphase chemistry mechanism system MCMv3.2/CAPRAM4.0/CAPRAM-DM1.0/CAPRAM-HM3.0 and the multiphase chemistry mechanism system with CAPRAM-DM1.0red and CAPRAM-HM3.0red. The CPU costs include gas- and aqueous-phase chemistry, microphysics, model initialization, and output.
2.5 Uncertainties of the new chemistry scheme
The downsizing of CAPRAM-HM3.0 and CAPRAM-DM1.0 solely, considering the most important reactions, has led to two new reduced mechanisms, which consist of reactions that sensitively impact the model outcome. Hence, the uncertainty of these reactions can be crucial for the model results. A discussion of the uncertainties of the mechanism development has already been performed in the previous papers describing the mechanism development of CAPRAM-HM2.0 (Bräuer et al., 2013), CAPRAM-HM3.0 (Hoffmann et al., 2019a, b), and CAPRAM-DM1.0 (Hoffmann et al., 2016) as well as in the cited laboratory work. That is why only a short discussion is given here.
For the oxidation of DMS in the gas phase, most of the rate constants are based on recommended values of the IUPAC database (http://iupac.pole-ether.fr/, last access: 20 April 2016) or Jet Propulsion Laboratory panel (JPL; Burkholder et al., 2015). Nevertheless, the application of the PSSA has modified the oxidation pathways, in particular the OH-addition reaction for DMS. The incorporation of the oxygen concentration might increase the uncertainty influencing the DMSO formation rate. As oxygen is in excess under tropospheric conditions and the oxygen concentration is treated in the new derived reaction rate constant specifically, minor changes are expectable. By contrast, no recommended values are available for the aqueous-phase reaction rate constants and can hence be stated as more uncertain (Hoffmann et al., 2016). Further laboratory work is required to minimize their uncertainties.
Regarding CAPRAM-HM3.0red, certain gas-phase reaction rate constants of the halogen chemistry are based on recommended values (Atkinson et al., 2006, 2007, 2008; Burkholder et al., 2015). However, for the oxidation of VOCs∕OVOCs by the Cl or Br atom, often only one reaction rate constant has been measured by laboratory studies. This is also true for many aqueous-phase chemistry reactions. The highest uncertainties are related to iodine chemistry. Here, often reaction rate constants, which might be of high atmospheric significance are based on estimations, only.
The CTM applied in this study is MUSCAT (Wolke et al., 2004, 2012). It is either coupled to the weather model COSMO (Consortium for Small-Scale Modelling; Steppeler et al., 2003; Baldauf et al., 2011) or ICON (ICOsahedral Non-hydrostatic; Zängl et al., 2015), which provide all required meteorological fields (e.g. wind, temperature, relative humidity, liquid water content, and precipitation) to MUSCAT that are necessary to calculate the advection, diffusion, and physico-chemical interaction of particles and trace gases. While the emission files of gases and aerosols within MUSCAT are generated by preprocessors, the chemical mechanism is imported from ASCII files, which allows for changes without code recompilation. In terms of dust (Heinold et al., 2007) and sea spray aerosols (Barthel et al., 2019), emissions can also be calculated online.
3.1 Implementation of CAPRAM-DM1.0red and CAPRAM-HM3.0red into COSMO-MUSCAT
Within the present study, the model framework COSMO-MUSCAT is used, which has recently been extended to be able to treat multiphase chemistry in clouds (Schrödner et al., 2014, 2018). The chemistry of DMS and detailed halogen chemistry are still missing. In order to consider them, the mechanisms CAPRAM-DM1.0red and CAPRAM-HM3.0red are implemented into the atmospheric multiphase chemistry core of COSMO-MUSCAT, in which gas-phase chemistry is described by the MOZART4 mechanism (Schultz et al., 2018) and aqueous-phase chemistry by the CAPRAM3.0red mechanism (Deguillaume et al., 2010). MOZART4 treats comprehensive halogen and DMS gas-phase chemistry that is replaced, as well as specific lumped VOCs and OVOCs. As outlined, these lumped species notably cover the VOCs and OVOCs, where the oxidation by the Cl atom is significant. To link CAPRAM-HM3.0red to MOZART4, the overall rate constants of the Cl atom are derived for the following lumped VOCs: (i) BIGALK, (ii) ALKOH, (iii) C2H5CHO, (iv) BIGALD1, (v) XYL, and (vi) BZALD. The lumped species C2H5CHO represents all aldehydes with more than three carbon atoms and is newly implemented in MOZART4, which makes it consistent with CAPRAM3.0red. Accordingly, the oxidation pathways are adjusted and the C2H5CHO oxidation by the OH radical and the Br atom has also been implemented.
Table 2 provides the implemented reaction rate constants of lumped VOCs. A full mechanistic description of the adjusted CAPRAM-DM1.0red and CAPRAM-HM3.0red is given in Tables S2–S10 in the Supplement.
Table 2Description of the lumped MOZART4 species and the corresponding kinetic reaction rate constants.

The recombination of halogenated peroxyl radicals as described above is adjusted to fit into MOZART4. In the MCM, this recombination is implemented for the sum of all RO2, whereas in MOZART4 it is often only for CH3O2 and CH3C(O)O2. The ratio of the MCM is applied in the RO2 recombination reaction, but RO2 is considered as CH3O2. The reaction rate constant is adopted from the corresponding CAPRAM-HM2.0 reaction (Bräuer et al., 2013).
Currently, the calculation of aqueous-phase chemical processes in MUSCAT is limited to cloudy conditions, i.e. a liquid water content (LWC) of above 0.01 g m−3. Furthermore, the chemical equilibria are treated dynamically as forward and backward reactions, which represents a critical challenge for the numerical solver. Deviations from the equilibrium state of rapid phase transfers and dissociations may lead to large chemical fluxes and hence small time steps, i.e. high computational costs. The robustness of the numerical integration is particularly affected at phase boundaries between cloud and non-cloud grid cells. Pre-balancing the aqueous-phase equilibria of CO2, NH3, HNO3, HCl, SO2, H2SO4, and organic acids in cloud-free grid cells at a predefined threshold LWC under cloud conditions (0.01 g m−3, abbreviated with “sub no. 1”) results in a robust numerical integration. This approach is similar to the pH calculation parameterization described by Alexander et al. (2012) but is designed to describe cloud chemistry and its effect on cloud droplet acidity in detail. In order to describe the activation of reactive halogen compounds by the chemistry in deliquesced aerosols, an additional sub-mechanism is introduced, assuming an LWC of 10 µg m−3 in cloud-free grid cells (abbreviated with “sub no. 2”). A schematic on how both sub-mechanisms enable the multiphase chemistry treatment in COSMO-MUSCAT can be seen in Fig. 3.
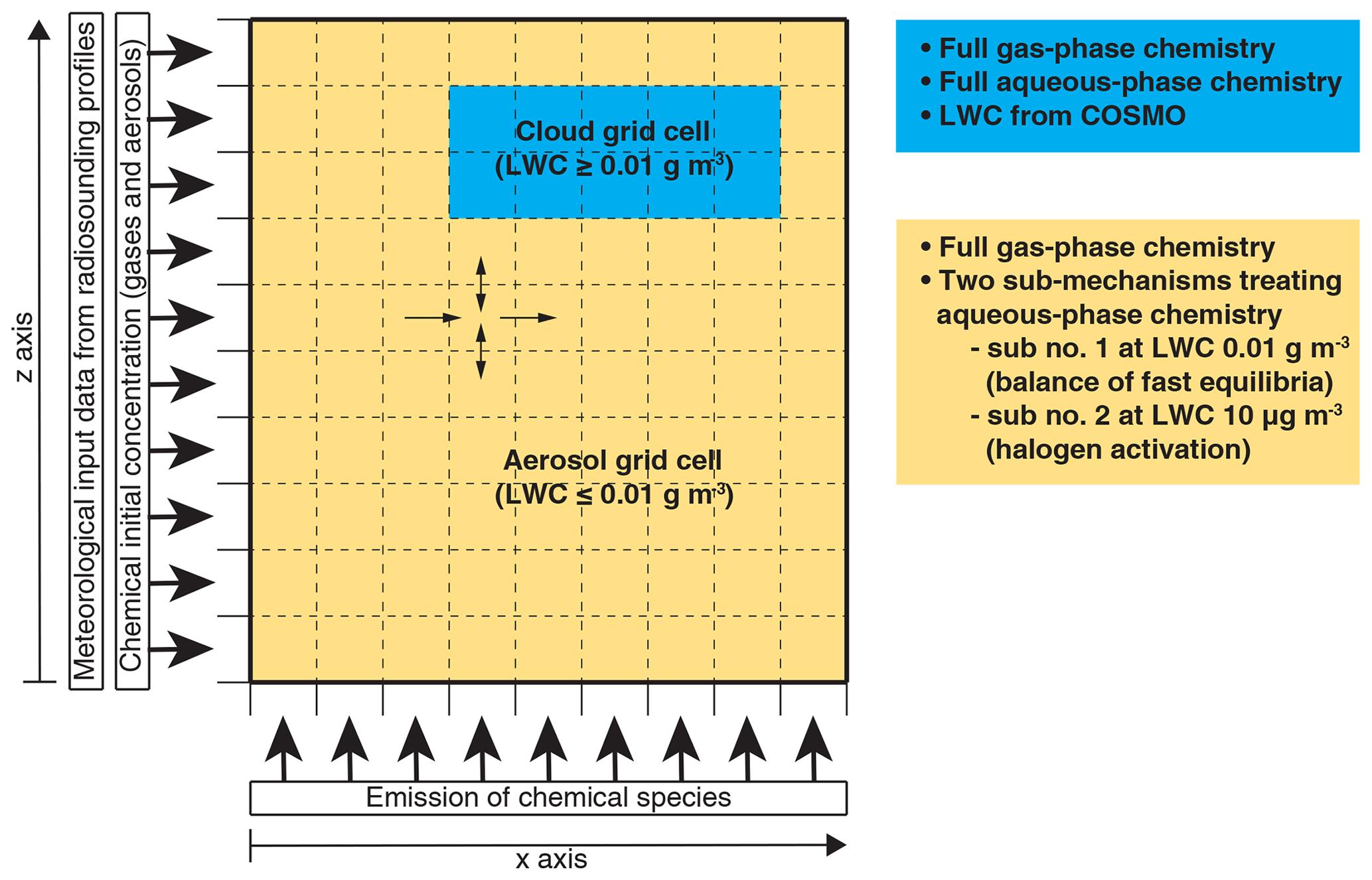
Figure 3Schematic representation of the multiphase chemistry treatment in COSMO-MUSCAT in an idealized 2D simulation.
The main halogen chemistry reactions treated in the “sub no. 2” sub-mechanism are the following:
The activation of halogens is in accordance with the heterogeneous reactions used in other CTMs (Badia et al., 2019; Hossaini et al., 2016; Jöckel et al., 2016; Long et al., 2014; Muniz-Unamunzaga et al., 2018; Saiz-Lopez et al., 2014; Wang et al., 2019). However, the “sub no. 2” sub-mechanism is able to treat pH-dependent processes, including (i) the activation of reactive halogen compounds and (ii) sulfite formation induced by HOX and H2O2 in the MBL online. Tables S8–S10 provide a complete overview of the treated aqueous-phase reactions and phase transfers in both the “sub no. 1” and the “sub no. 2” sub-mechanism in CAPRAM-DM1.0red and CAPRAM-HM3.0red. Overall, COSMO-MUSCAT now represents the CTM with the most detailed description of marine multiphase chemistry (see Fig. 4).
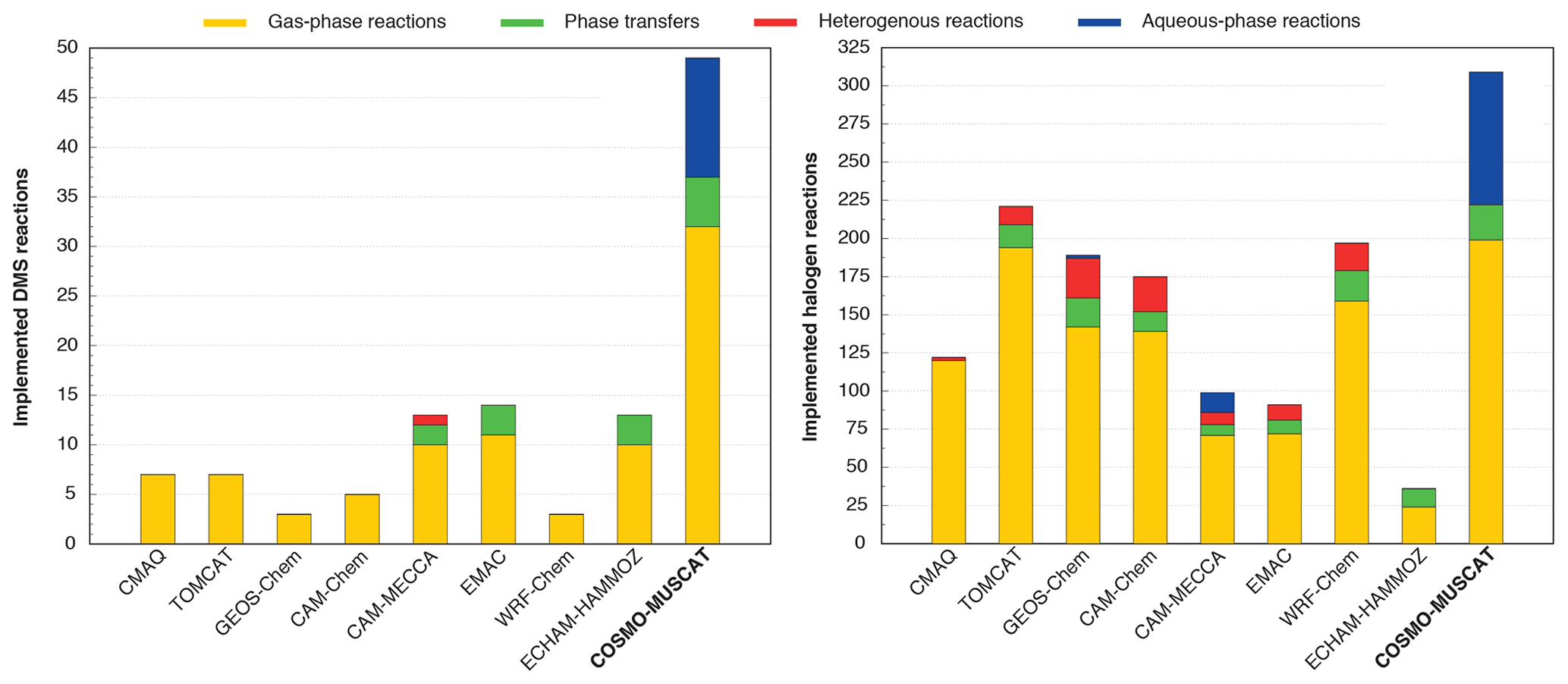
Figure 4Comparison of applied tropospheric DMS and halogen chemistry mechanisms within the chemical transport models: CMAQ (Muniz-Unamunzaga et al., 2018), TOMCAT (Hossaini et al., 2016), GEOS-Chem (Wang et al., 2019), CAM-Chem (Saiz-Lopez et al., 2014), CAM-MECCA (Long et al., 2014), EMAC (Jöckel et al., 2016), and WRF-Chem (Badia et al., 2019). EMAC is a chemistry climate model that can be run as a CTM.
3.2 Evaluation of the 2D implementation
The evaluation of the implementation is carried out by two 2D simulations (x–z cross section) dealing with a pristine ocean scenario under two meteorological conditions, namely a convective and stable atmospheric layer, which will result in modelled convective and stratiform clouds, respectively. Both scenarios are further applied to investigate DMS oxidation in more detail. The evaluation of the implementation is performed by investigating the activation of halogen compounds in the MBL and comparing it with available ambient measurements and other model data. 2D simulations are preferred over 3D ones, because they are based on the same meteorological dynamics but have one lower degree of freedom. As a result, the system under investigation is less computationally expensive. Additionally, 2D simulations enable a comprehensive understanding of multiphase chemistry in the atmospheric column, including vertical mixing processes.
3.2.1 2D model setup
The chemical model setup, i.e. the initialization, deposition, and emission of trace gases and VOCs, as well as the aerosol composition to describe the atmospheric composition of the pristine ocean, is the same as the one used during the mechanism reduction. Because of the lumped species within MOZART4, specific VOC emissions are merged. Table S1 provides the emission rates. Initialized concentrations of gas-phase and aerosol compounds represent ground values and are distributed vertically as constant mass mixing ratios within the model domain of MUSCAT. These values are used as constant boundary conditions on the left-hand side of the model domain (see Fig. 3).
Two different meteorological cases are simulated: one with a high diversity of clouds and a strong vertical wind velocity (further called unstable meteorological condition) and another with a stable cloud cover at the top of the marine boundary layer and a weak vertical wind velocity (further called stable meteorological condition). These two different meteorological pristine ocean simulations are chosen to evaluate the numerical robustness of the model. The meteorological scenarios are initialized using radiosonde profiles (unstable meteorological condition station: Camborne Observations, station identifier: 3808 on 12 Z 21 June 2016 and stable meteorological condition station: GVAC Sal Observations, station identifier: 8594 on 12 Z 12 June 2017), which can be read and processed by COSMO by default and are considered constant meteorological boundary conditions on the left-hand side of the model domain (see Fig. 3).
The model domain spans 400 horizontal grid cells with a resolution of 1.11 km per grid cell and 100 vertical levels with a resolution of 100 m. Whereas COSMO is run on the full domain, only the inner 200 horizontal grid cells (overall 222 km) and lowermost 15 vertical levels (overall 1500 m) are used for the multiphase chemistry simulations with MUSCAT. MUSCAT uses a smaller domain as the interaction is sufficient to describe the multiphase chemistry in the marine boundary layer (MBL). Furthermore, the height of the MBL is often lower than 1000 m (Norris, 1998; Carrillo et al., 2016), which enables an investigation of the interactions between the MBL and the free troposphere and significantly saves computation time compared to the full domain. So, this chosen model setup can capture almost all essential chemical processes as well as the distribution of important species in the marine troposphere. Overall, the modelling domain for multiphase chemistry encompasses 3000 grid cells.
3.2.2 Comparison with measurements
HCl gas-phase concentration
Firstly, the modelled concentration range of gaseous HCl is compared to actual measurements. Figure 5 shows that very high HCl concentrations are modelled, especially in the stable meteorological condition simulation above the MBL. These are results of the constant vertical distributed mass mixing ratios of the initial values. Since the evaluation focuses on the activation of HCl within the MBL, these values can be neglected. The modelled values below 1000 m outside of clouds are of the order of 109 molecules cm−3 after 12 h of modelling time and thus in the range of both other modelled (Hossaini et al., 2016; Wang et al., 2019) and measured values within the marine pristine boundary layer (e.g. Keene and Savoie, 1999; Sander et al., 2013; Pszenny et al., 2004). The higher LWC of clouds results in strongly reduced HCl gas-phase concentrations due to the phase partitioning shift from the gas to the aqueous phase. Further chemical cloud processing increases aerosol acidity, yielding higher HCl gas-phase concentrations behind the cloud in wind direction (see Fig. 5).
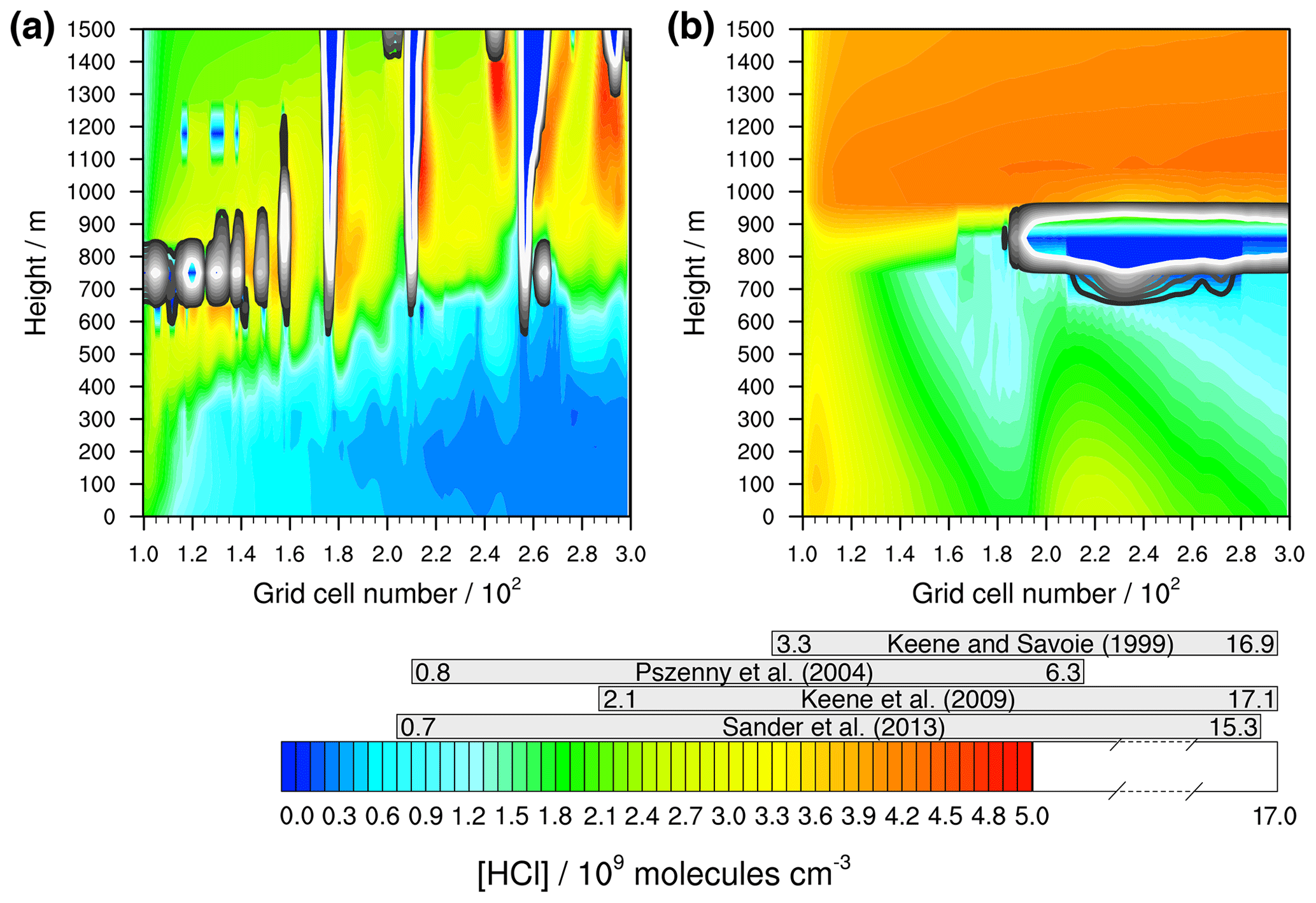
Figure 5Simulated concentrations of HCl in the gas phase by COSMO-MUSCAT (a) in the unstable meteorological condition simulation with convective clouds and (b) in the stable meteorological condition simulation with stratiform clouds after 12 h of modelling time. The x axis represents the innermost horizontal grid cells divided by 100. The grey bars represent measured values in most likely pristine marine environments (see Table S11 for further details). The black contour lines represent the simulated clouds. The black line corresponds to a LWC of 0.01 g m−3 and the white line to 0.1 g m−3. The area framed by the white line includes LWC above 0.1 g m−3.
BrO gas-phase concentration
Contrary to HCl, the measured concentrations of HBr over the pristine ocean are missing. However, a high number of measured gas-phase BrO concentrations in the MBL are available (Saiz-Lopez and von Glasow, 2012; Simpson et al., 2015). Therefore, as a second step, the modelled gas-phase BrO concentration range is compared to measurements. Figure 6 shows that the modelled values outside of the cloud grid cells ranging from 106 to 107 molecules cm−3 after 12 h of modelling time. Thus, they are in the range of other modelled (Zhu et al., 2019) as well as measured values within the pristine MBL (e.g. Leser et al., 2003; Read et al., 2008; Chen et al., 2016). Apart from that, the vertical distribution significantly differs between both simulations resulting into distinct spatial pattern. At the left-hand side of the model domain, the BrO concentration is similar, which is related to the activation of reactive bromine species from the initialized marine aerosols. However, when clouds are formed the profiles change. This is related to the high differences in the vertical wind field (see Fig. 7a and b). Because of the stronger updraughts in the unstable meteorological condition simulation, the reactive halogen compounds are advected towards higher altitudes compared to the slow vertical winds in the stable meteorological condition simulation. A second remarkable difference is the much lower BrO concentration at the right-hand side of the model domain in the stable meteorological condition simulation. This effect is more explicitly discussed in Sect. 3.3.3.

Figure 6Simulated concentrations of bromine monoxide in the gas phase by COSMO-MUSCAT (a) in the unstable meteorological condition simulation with convective clouds and (b) in the stable meteorological condition simulation with stratiform clouds after 12 h of modelling time. The x axis represents the innermost horizontal grid cells divided by 100. The grey bars represent measured values in most likely pristine marine environments (see Table S11 for further details). The black contour lines represent the simulated clouds. The black line corresponds to a LWC of 0.01 g m−3 and the white line to 0.1 g m−3. The area framed by the white line includes LWC above 0.1 g m−3.
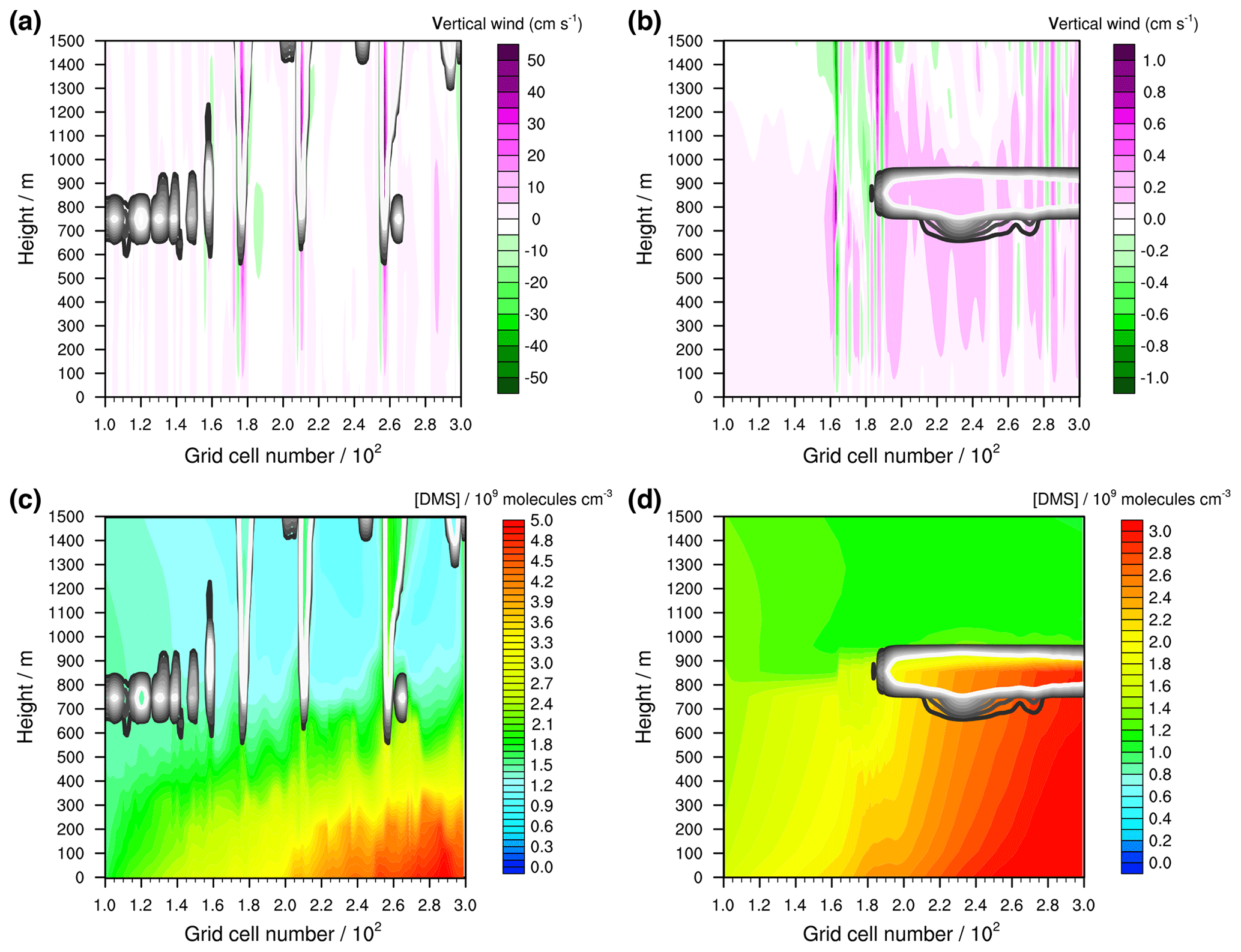
Figure 7Modelled vertical winds (cm s−1) (a) in the unstable meteorological condition simulation with convective clouds and (b) in the stable meteorological condition simulation with stratiform clouds after 12 h of modelling time. The x axis represents the innermost horizontal grid cells divided by 100. The black contour lines represent the simulated clouds. The black line corresponds to a LWC of 0.01 g m−3 and the white line to 0.1 g m−3. The area framed by the white line includes LWC above 0.1 g m−3. Further, the modelled concentrations of DMS in the gas phase (109 molecules cm−3) are shown in (c) for the unstable meteorological condition simulation with convective clouds and in (d) for the stable meteorological condition simulation with stratiform clouds after 12 h of modelling time.
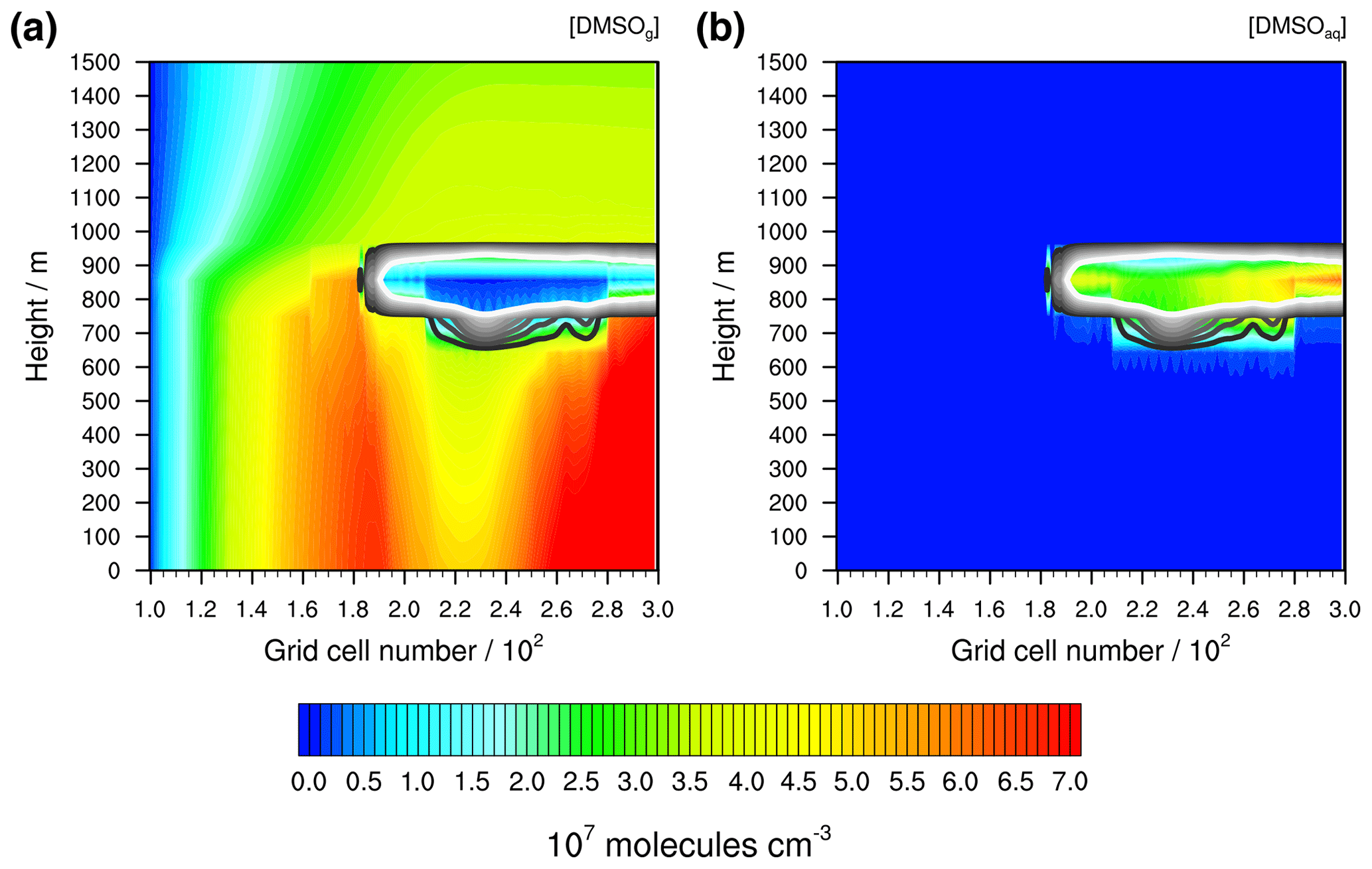
Figure 8Simulated concentrations of DMSO (a) in the gas phase and (b) in the aqueous phase under the stable meteorological condition simulation with stratiform clouds after 12 h of modelling time. The x axis represents the innermost horizontal grid cells divided by 100. The black contour lines represent the simulated clouds. The black line corresponds to a LWC of 0.01 g m−3 and the white line to 0.1 g m−3. The area framed by the white line includes LWC above 0.1 g m−3.
Apart from that, many field studies had the problem that the BrO concentration in the MBL was always below the detection limit (Sander et al., 2003). In a recent measurement study, the measured BrO in the MBL was also always below the detection limit of 0.5 pptv, i.e. around 1.2×107 molecules cm−3 (Volkamer et al., 2015). Hence, the BrO concentrations are modelled adequately. Since the activation of reactive bromine is highly related to that of chlorine, the mechanism is able to represent the activation of reactive halogen compounds within the MBL. A comparison of reactive iodine compounds with measurements is not performed, because the concentration range is highly sensitive to the chosen emission values of molecular iodine and iodinated organics from the sea surface and is thus uncertain.
Overall, the new marine multiphase chemistry model can represent marine aerosol chemistry and linked halogen activation under consideration of meteorological dynamics and shows a good agreement to other field as well as model data. Thus, it is applicable for further detailed 3D studies.
3.3 Results of pristine ocean scenarios
3.3.1 Vertical wind and DMS distribution
Both scenarios are further applied to investigate the multiphase oxidation pathways of DMS in a cloudy marine atmosphere in detail. Figure 7 shows the modelled distribution of clouds and the strength of the vertical wind field as well as the modelled DMS concentration distribution for both simulations after 12 h of modelling time. In the unstable meteorological condition simulation, the clouds extend up to a height of more than 2000 m, whereas in the stable meteorological condition simulation, the top of the cloud is capped below an inversion layer at around 1000 m. Also, the vertical winds are much stronger in the unstable meteorological condition simulation. Because of the strong vertical winds, gas-phase DMS concentrations of around 2×109 molecules cm−3 are transported into the lower free troposphere. The strong inversion and low magnitude of the vertical wind speed in the stable meteorological condition simulation hinders effective DMS transportation into the free troposphere, resulting in a DMS concentration of around 1×109 molecules cm−3 above the MBL. This is consistent with the initialized background DMS concentration. Below the inversion, DMS concentrations are more homogeneously distributed, which is different to the unstable meteorological condition simulation, with a stronger variability related to the vertical wind field, i.e. the peaking of DMS concentrations into higher vertical levels because of strong updraughts (cp. Fig. 7).
3.3.2 Vertical DMSO distribution
If such homogeneously distributed DMS concentrations are modelled as in the stable meteorological condition simulation in a clear sky atmosphere, a similar concentration distribution for the first stable DMS oxidation products will be modelled. Hence, the concentration distribution of DMSO is a good indicator to investigate the effect of clouds on DMS oxidation. In Fig. 8, the distribution of DMSO in the gas and aqueous phases in the stable meteorological condition simulation after 12 h of modelling time is shown. Furthermore, the overall modelled DMSO production and loss rates separated into gas- and aqueous-phase reactions were added to the Supplement (see Fig. S11).
The stratiform clouds have a very high influence on the DMSO concentration in both the gas and the aqueous phases. The spatial gas-phase DMSO concentration distribution differs from the DMS concentration. Below the optically thickest clouds, the gas-phase DMSO concentration is significantly reduced, whereas above the cloud it slightly increases. In the cloud grid cells, the gas-phase DMSO concentration is reduced significantly because of the uptake into the aqueous phase. The reduced gas-phase concentrations below the cloud cannot be explained by vertical or horizontal transportation, because, as can be seen in Fig. 7, an updraught would result in observable concentration peaks in the higher vertical levels. Therefore, the gas-phase DMSO formation in the MBL is somehow influenced by the cloud above, which makes it necessary to investigate the cloud-induced effect on such crucial DMS oxidants in the pristine MBL.

Figure 9Simulated oxidation rate of DMS by BrO in the stable meteorological condition simulation with stratiform clouds after 12 h of modelling time. The x axis represents the innermost horizontal grid cells divided by 100. The black contour lines represent the simulated clouds. The black line corresponds to a LWC of 0.01 g m−3 and the white line to 0.1 g m−3. The area framed by the white line includes LWC above 0.1 g m−3.
3.3.3 Effects of stratiform clouds on DMS oxidation
Within the pristine MBL, the BrO radical is a primary DMS oxidant that forms DMSO (Barnes et al., 2006; Breider et al., 2010; Hoffmann et al., 2016; von Glasow and Crutzen, 2004; Chen et al., 2018). This radical is formed through reaction of O3 by the Br atom that is activated by multiphase chemistry. The following reactions are important pathways of activation in a clear sky MBL (von Glasow and Crutzen, 2004; von Glasow et al., 2002b):
In the pristine marine boundary layer, two competing pathways determine the main fate of the BrO radical through (i) a reaction with DMS and (ii) a reaction with HO2. The oxidation of DMS leads to DMSO and the Br atom, so that a cycle is established that continuously depletes O3 and forms DMSO as long as DMS is emitted or ozone is available. This cycle is disturbed by the reaction of BrO with HO2, yielding HOBr, which can be photolysed back into the Br atom again or converted by multiphase chemistry into BrCl or Br2. Overall, the photolysis of HOBr, Br2 and BrCl determine the DMS to DMSO conversion. Clouds suppress the photolysis of Br2, BrCl, and HOBr due to the reflection of incoming solar radiation. The thicker the cloud, the lower the radiation flux below. Consequently, Br atom activation below the cloud is hindered, affecting the BrO concentration and thus the reaction rate of BrO with DMS that yields DMSO (see Fig. 9). Due to a longer lifetime against further oxidation and corresponding horizontal advection, the DMSO concentration profile is shifted towards the right compared to BrO. The lowest oxidation flux between DMS and BrO is modelled between grid cell 2.0 and 2.15. The effect on DMSO concentration is modelled between grid cell 2.1 and 2.4.
The photolysis of BrCl and Br2 is highly sensitive to cloud shading and thus has a high impact on the formation of reactive bromine and the linked DMS oxidation. Moreover, model studies suggest that BrCl photolysis is an important contributor to Cl atom activation in the MBL (Wang et al., 2019; von Glasow et al., 2002b). Hence, the outlined model results reveal that the shading effect of clouds is also very important for the atmospheric Cl atom concentration budget, affecting the atmospheric oxidation capacity within the MBL.
3.3.4 Formation of MSA and aqueous sulfate
DMSO is rapidly oxidized into MSIA and thus a similar MSIA profile is modelled. As MSIA is highly reactive in the gas and aqueous phases as well as highly soluble, it is rapidly oxidized into methane sulfonate (MS−) in both the aerosol and the cloud phases. There, O3 is the preferred oxidant in the aerosol phase, whereas in cloud droplets it is the OH radical (Hoffmann et al., 2016). The MS− formed in cloud can be transported towards the ground by downdraughts. However, comparing the DMS concentrations in Fig. 7 with the DMSO concentrations in Fig. 8, the up- and downdraughts in the stable meteorological condition simulations have little effect on the concentration distribution in height. The strongest effect relates to the advection from the left-hand to the right-hand side of the model domain and continuous emission from the surface. In the grid cells left of the cloud, the DMSO concentration is high and consequently the aerosol particle chemistry of DMSO and of the subsequent oxidation product MSIA leads to a sharp increase of MS− formation at the grid cells below the left cloud edge (see Fig. 10a). Due to the advection of the stable MS− to the right-hand side of the model domain, the spatial profiles of DMSO (Fig. 8) and MS− differ. The high modelled chemical fluxes in cloud droplets indicate the highest MS− concentrations to be within and below the cloud grid cells.
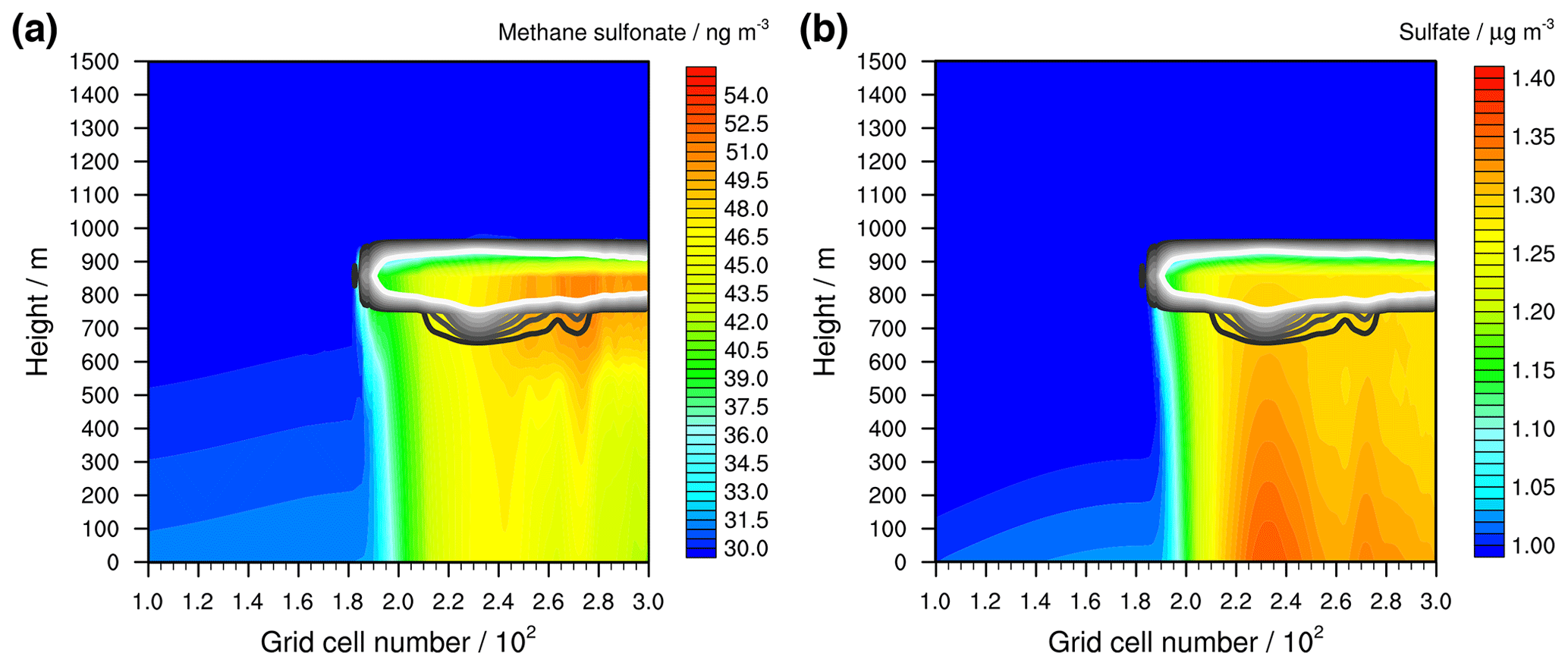
Figure 10Simulated aqueous-phase concentrations of (a) methane sulfonate and (b) sulfate in the stable meteorological condition simulation with stratiform clouds after 12 h of modelling time. The x axis represents the innermost horizontal grid cells divided by 100. The black contour lines represent the simulated clouds. The black line corresponds to a LWC of 0.01 g m−3 and the white line to 0.1 g m−3. The area framed by the white line includes LWC above 0.1 g m−3. The initial background concentration of methane sulfonate is at about 30 ng m−3 and that of sulfate at 1 µg m3.
Also, the concentration of sulfate (see Fig. 10b) is enhanced in the grid cells at the left cloud edge but because of different reasons. At the left cloud edge, the lower photolysis rates increase the SO2 oxidation into sulfate by HOX and H2O2. Hence, a stronger HOX-related (especially HOI) reactive SO2 uptake on the aerosols is modelled. The reaction of HOBr results in the formation of bromide. In addition to the uptake of HBr, this increases the bromide concentration in cloud droplets by up to 1 order of magnitude compared to the ground-level concentration before the left cloud edge (see Fig. S12). The uptake is highest under the optically thickest modelled clouds, resulting in the highest modelled sulfate concentrations. Therefore, the modelled spatial concentration is contrary to that of DMSO.
Reduced multiphase chemistry mechanisms of DMS and reactive halogen compounds are developed through the reduction of the near-explicit multiphase chemistry mechanisms CAPRAM-DM1.0 and CAPRAM-HM3.0. Simulations that compare the reduced with the original mechanisms revealed that the reduced mechanisms are able to reproduce the concentrations and time evolutions of main air pollutants as well as key reactive halogen compounds. Additionally, CPU time in the box model simulations is reduced by 16 %, 5 %, and 6 %, depending on the model scenario. Afterwards, the reduced mechanisms are implemented into the chemistry transport model COSMO-MUSCAT. This process is evaluated by idealized 2D simulations of an atmospheric pristine ocean environment. It was proven that the reduced marine multiphase chemistry mechanism can represent marine aerosol chemistry and linked halogen activation as it matches measured field concentrations, e.g. HCl and BrO.
Following that implementation, 2D simulations of a pristine ocean scenario are carried out, investigating the effect of stable (stratiform cloud) and more unstable meteorological conditions (convective clouds) on multiphase DMS oxidation. The simulations reveal that clouds have both strong direct and indirect photochemical effects on the oxidation and vertical distribution of DMS in the marine atmosphere. Firstly, locally high updraught velocities in the unstable scenario result in fast transport of DMS from the marine boundary layer into the free troposphere. Hence, transport and further oxidation of DMS can be an important source of SO2 within the free troposphere, particularly in the Southern Ocean region that is less affected by anthropogenic pollution. Secondly, clouds enhance the formation of MSA via the DMS addition channel. The formed DMSO is effectively consumed by cloud droplets, where it is rapidly oxidized into MSA. Thirdly, the shading of clouds has a high impact on the photolysis of dihalogens that are the main contributor to Cl and Br atom activation. Thus, a much lower oxidation of DMS into DMSO occurs below stratiform clouds. In contrast, the lower HOX photolysis induces stronger sulfate formation. The results indicate that clouds strongly affect the oxidation of DMS directly because of enhanced aqueous-phase oxidation into MSA and indirectly by suppressing the DMSO formation due to lower halogen atom activation. In total, a strong possible effect on the atmospheric oxidation capacity within the MBL of the pristine ocean is assumed. The important effect of wet scavenging by clouds was not investigated as COSMO-MUSCAT(5.04e) did not implement it in detail but represented it using a first-order scavenging rate. Future studies aim to implement a more precise scheme. Since the clouds modelled in this study are not known to precipitate, the propagated error should be small.
Overall, the 2D simulations demonstrate the capability of COSMO-MUSCAT to now cover the multiphase chemistry in marine-influenced atmospheric environments. This allows for deeper investigations of multiphase chemistry in a wide range of temporal and spatial resolutions together with transport and microphysical processes in the future. In a next step, the mechanism will be applied in simulations with COSMO-MUSCAT for modelling measurement campaigns at the Cape Verde Atmospheric Observatory (Carpenter et al., 2010), supporting the interpretation of the measurement data and enabling further model and/or mechanism evaluation. Finally, the reduced mechanism is designed in such a way that new findings in DMS or halogen chemistry can easily be implemented, e.g. improved understanding of the multiphase chemistry of the unimolecular H-shift of CH3SCH2O2.
The code for the COSMO model is available according to the Software License Agreement by Deutscher Wetterdienst (German Weather Service, http://cosmo-model.org, last access: 12 November 2019). The source code of MUSCAT and SPACCIM, external parameters, and applied mechanisms are archived on a local Git server and can be obtained by request through Ralf Wolke (wolke@tropos.de). Access to the model code used in the paper has been granted to the editor.
The supplement related to this article is available online at: https://doi.org/10.5194/gmd-13-2587-2020-supplement.
EHH, AT, and RW did the model development on SPACCIM. EHH, AT, and HH designed the SPACCIM modelling work. EHH performed the SPACCIM simulations. EHH, AT, and HH analysed the SPACCIM model results. EHH, RS, and RW did the model development on COSMO-MUSCAT. EHH, RS, and RW designed the COSMO-MUSCAT modelling work. EHH performed the COSMO-MUSCAT simulations. EHH analysed the COSMO-MUSCAT model results. EHH, RS, AT, and HH wrote the paper.
The authors declare that they have no conflict of interest.
Erik H. Hoffmann thanks the PhD scholarship programme of the German Federal Environmental Foundation (Deutsche Bundesstiftung Umwelt, DBU, AZ: 2016/424) for its financial support. This work has received funding from the European Union's Horizon 2020 research and innovation programme through the EUROCHAMP-2020 Infrastructure Activity under grant agreement no. 730997.
This research has been supported by the European Commission (EUROCHAMP-2020 (grant no. 730997)). This work was also supported by the EU Marie Skłodowska-Curie Actions (grant no. 690958-MARSU-RISE-2015).
This paper was edited by Andrea Stenke and reviewed by two anonymous referees.
Alexander, B., Allman, D. J., Amos, H. M., Fairlie, T. D., Dachs, J., Hegg, D. A., and Sletten, R. S.: Isotopic constraints on the formation pathways of sulfate aerosol in the marine boundary layer of the subtropical northeast Atlantic Ocean, J. Geophys. Res.-Atmos., 117, D06304, https://doi.org/10.1029/2011jd016773, 2012.
Ammann, M., Cox, R. A., Crowley, J. N., Jenkin, M. E., Mellouki, A., Rossi, M. J., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume VI – heterogeneous reactions with liquid substrates, Atmos. Chem. Phys., 13, 8045–8228, https://doi.org/10.5194/acp-13-8045-2013, 2013.
Andreae, M. O.: Ocean-atmosphere interactions in the global biogeochemical sulfur cycle, Mar. Chem., 30, 1–29, https://doi.org/10.1016/0304-4203(90)90059-L, 1990.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume III – gas phase reactions of inorganic halogens, Atmos. Chem. Phys., 7, 981–1191, https://doi.org/10.5194/acp-7-981-2007, 2007.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume IV – gas phase reactions of organic halogen species, Atmos. Chem. Phys., 8, 4141–4496, https://doi.org/10.5194/acp-8-4141-2008, 2008.
Badia, A., Reeves, C. E., Baker, A. R., Saiz-Lopez, A., Volkamer, R., Koenig, T. K., Apel, E. C., Hornbrook, R. S., Carpenter, L. J., Andrews, S. J., Sherwen, T., and von Glasow, R.: Importance of reactive halogens in the tropical marine atmosphere: a regional modelling study using WRF-Chem, Atmos. Chem. Phys., 19, 3161–3189, https://doi.org/10.5194/acp-19-3161-2019, 2019.
Baklanov, A., Schlünzen, K., Suppan, P., Baldasano, J., Brunner, D., Aksoyoglu, S., Carmichael, G., Douros, J., Flemming, J., Forkel, R., Galmarini, S., Gauss, M., Grell, G., Hirtl, M., Joffre, S., Jorba, O., Kaas, E., Kaasik, M., Kallos, G., Kong, X., Korsholm, U., Kurganskiy, A., Kushta, J., Lohmann, U., Mahura, A., Manders-Groot, A., Maurizi, A., Moussiopoulos, N., Rao, S. T., Savage, N., Seigneur, C., Sokhi, R. S., Solazzo, E., Solomos, S., Sørensen, B., Tsegas, G., Vignati, E., Vogel, B., and Zhang, Y.: Online coupled regional meteorology chemistry models in Europe: current status and prospects, Atmos. Chem. Phys., 14, 317–398, https://doi.org/10.5194/acp-14-317-2014, 2014.
Baldauf, M., Seifert, A., Förstner, J., Majewski, D., Raschendorfer, M., and Reinhardt, T.: Operational Convective-Scale Numerical Weather Prediction with the COSMO Model: Description and Sensitivities, Mon. Weather Rev., 139, 3887–3905, https://doi.org/10.1175/mwr-d-10-05013.1, 2011.
Barnes, I., Hjorth, J., and Mihalopoulos, N.: Dimethyl sulfide and dimethyl sulfoxide and their oxidation in the atmosphere, Chem. Rev., 106, 940–975, https://doi.org/10.1021/Cr020529+, 2006.
Barthel, S., Tegen, I., and Wolke, R.: Do new sea spray aerosol source functions improve the results of a regional aerosol model?, Atmos. Environ., 198, 265–278, https://doi.org/10.1016/j.atmosenv.2018.10.016, 2019.
Benedict, K. B., Lee, T., and Collett, J. L.: Cloud water composition over the southeastern Pacific Ocean during the VOCALS regional experiment, Atmos. Environ., 46, 104–114, https://doi.org/10.1016/j.atmosenv.2011.10.029, 2012.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Hoffmann, E. H., Tilgner, A., Hyttinen, N., Prisle, N. L., Hansel, A., and Herrmann, H.: Fast Peroxy Radical Isomerization and OH Recycling in the Reaction of OH Radicals with Dimethyl Sulfide, J. Phys. Chem. Lett., 10, 6478–6483, https://doi.org/10.1021/acs.jpclett.9b02567, 2019.
Berresheim, H., Huey, J. W., Thorn, R. P., Eisele, F. L., Tanner, D. J., and Jefferson, A.: Measurements of dimethyl sulfide, dimethyl sulfoxide, dimethyl sulfone, and aerosol ions at Palmer Station, Antarctica, J. Geophys. Res.-Atmos., 103, 1629–1637, https://doi.org/10.1029/97JD00695, 1998.
Bräuer, P., Tilgner, A., Wolke, R., and Herrmann, H.: Mechanism development and modelling of tropospheric multiphase halogen chemistry: The CAPRAM Halogen Module 2.0 (HM2), J. Atmos. Chem., 70, 19–52, https://doi.org/10.1007/s10874-013-9249-6, 2013.
Bräuer, P., Mouchel-Vallon, C., Tilgner, A., Mutzel, A., Böge, O., Rodigast, M., Poulain, L., van Pinxteren, D., Wolke, R., Aumont, B., and Herrmann, H.: Development of a protocol for the auto-generation of explicit aqueous-phase oxidation schemes of organic compounds, Atmos. Chem. Phys., 19, 9209–9239, https://doi.org/10.5194/acp-19-9209-2019, 2019.
Breider, T. J., Chipperfield, M. P., Richards, N. A. D., Carslaw, K. S., Mann, G. W., and Spracklen, D. V.: Impact of BrO on dimethylsulfide in the remote marine boundary layer, Geophys. Res. Lett., 37, L02807, https://doi.org/10.1029/2009gl040868, 2010.
Brown, S. S. and Stutz, J.: Nighttime radical observations and chemistry, Chem. Soc. Rev., 41, 6405–6447, https://doi.org/10.1039/c2cs35181a, 2012.
Burkholder, J. B., Sander, S. P., Abbatt, J., Barker, J. R., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Wilmouth, D. M., and Wine, P. H.: Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 18, Jet Propulsion Laboratory, Pasadena, 2015.
Carpenter, L. J. and Nightingale, P. D.: Chemistry and release of gases from the surface ocean, Chem. Rev., 115, 4015–4034, https://doi.org/10.1021/cr5007123, 2015.
Carpenter, L. J., Fleming, Z. L., Read, K. A., Lee, J. D., Moller, S. J., Hopkins, J. R., Purvis, R. M., Lewis, A. C., Müller, K., Heinold, B., Herrmann, H., Fomba, K. W., van Pinxteren, D., Müller, C., Tegen, I., Wiedensohler, A., Müller, T., Niedermeier, N., Achterberg, E. P., Patey, M. D., Kozlova, E. A., Heimann, M., Heard, D. E., Plane, J. M. C., Mahajan, A., Oetjen, H., Ingham, T., Stone, D., Whalley, L. K., Evans, M. J., Pilling, M. J., Leigh, R. J., Monks, P. S., Karunaharan, A., Vaughan, S., Arnold, S. R., Tschritter, J., Pöhler, D., Frieß, U., Holla, R., Mendes, L. M., Lopez, H., Faria, B., Manning, A. J., and Wallace, D. W. R.: Seasonal characteristics of tropical marine boundary layer air measured at the Cape Verde Atmospheric Observatory, J. Atmos. Chem., 67, 87–140, https://doi.org/10.1007/s10874-011-9206-1, 2010.
Carpenter, L. J., Archer, S. D., and Beale, R.: Ocean-atmosphere trace gas exchange, Chem. Soc. Rev., 41, 6473–6506, https://doi.org/10.1039/c2cs35121h, 2012.
Carpenter, L. J., MacDonald, S. M., Shaw, M. D., Kumar, R., Saunders, R. W., Parthipan, R., Wilson, J., and Plane, J. M. C.: Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine, Nat. Geosci., 6, 108–111, https://doi.org/10.1038/ngeo1687, 2013.
Carrillo, J., Guerra, J. C., Cuevas, E., and Barrancos, J.: Characterization of the Marine Boundary Layer and the Trade-Wind Inversion over the Sub-tropical North Atlantic, Bound.-Lay. Meteorol., 158, 311–330, https://doi.org/10.1007/s10546-015-0081-1, 2016.
Charlson, R. J., Lovelock, J. E., Andreae, M. O., and Warren, S. G.: Oceanic phytoplankton, atmospheric sulfur, cloud albedo and climate, Nature, 326, 655–661, https://doi.org/10.1038/326655a0, 1987.
Chen, D., Huey, L. G., Tanner, D. J., Salawitch, R. J., Anderson, D. C., Wales, P. A., Pan, L. L., Atlas, E. L., Hornbrook, R. S., Apel, E. C., Blake, N. J., Campos, T. L., Donets, V., Flocke, F. M., Hall, S. R., Hanisco, T. F., Hills, A. J., Honomichl, S. B., Jensen, J. B., Kaser, L., Montzka, D. D., Nicely, J. M., Reeves, J. M., Riemer, D. D., Schauffler, S. M., Ullmann, K., Weinheimer, A. J., and Wolfe, G. M.: Airborne measurements of BrO and the sum of HOBr and Br2 over the Tropical West Pacific from 1 to 15 km during the CONvective TRansport of Active Species in the Tropics (CONTRAST) experiment, J. Geophys. Res.-Atmos., 121, 12560–12578, https://doi.org/10.1002/2016JD025561, 2016.
Chen, Q., Schmidt, J. A., Shah, V., Jaegle, L., Sherwen, T., and Alexander, B.: Sulfate production by reactive bromine: Implications for the global sulfur and reactive bromine budgets, Geophys. Res. Lett., 44, 7069–7078, https://doi.org/10.1002/2017gl073812, 2017.
Chen, Q., Sherwen, T., Evans, M., and Alexander, B.: DMS oxidation and sulfur aerosol formation in the marine troposphere: a focus on reactive halogen and multiphase chemistry, Atmos. Chem. Phys., 18, 13617–13637, https://doi.org/10.5194/acp-18-13617-2018, 2018.
Davis, D., Chen, G., Kasibhatla, P., Jefferson, A., Tanner, D., Eisele, F., Lenschow, D., Neff, W., and Berresheim, H.: DMS oxidation in the Antarctic marine boundary layer: Comparison of model simulations and held observations of DMS, DMSO, DMSO2, H2SO4(g), MSA(g), and MSA(p), J. Geophys. Res.-Atmos., 103, 1657–1678, https://doi.org/10.1029/97JD03452, 1998.
Deguillaume, L., Tilgner, A., Schrödner, R., Wolke, R., Chaumerliac, N., and Herrmann, H.: Towards an operational aqueous phase chemistry mechanism for regional chemistry-transport models: CAPRAM-RED and its application to the COSMO-MUSCAT model, J. Atmos. Chem., 64, 1–35, https://doi.org/10.1007/s10874-010-9168-8, 2010.
Ervens, B., George, C., Williams, J. E., Buxton, G. V., Salmon, G. A., Bydder, M., Wilkinson, F., Dentener, F., Mirabel, P., and Herrmann, H.: CAPRAM 2.4 (MODAC mechanism): An extended and condensed tropospheric aqueous phase mechanism and its application, J. Geophys. Res., 108, 4426, https://doi.org/10.1029/2002jd002202, 2003.
Falbe-Hansen, H., Sørensen, S., Jensen, N. R., Pedersen, T., and Hjorth, J.: Atmospheric gas-phase reactions of dimethylsulphoxide and dimethylsulphone with OH and NO3 radicals, Cl atoms and ozone, Atmos. Environ., 34, 1543–1551, https://doi.org/10.1016/S1352-2310(99)00407-0, 2000.
Farmer, D. K., Cappa, C. D., and Kreidenweis, S. M.: Atmospheric processes and their controlling influence on cloud condensation nuclei activity, Chem. Rev., 115, 4199–4217, https://doi.org/10.1021/cr5006292, 2015.
Faxon, C. B. and Allen, D. T.: Chlorine chemistry in urban atmospheres: a review, Environ. Chem., 10, 221–233, https://doi.org/10.1071/en13026, 2013.
Gaston, C. J., Riedel, T. P., Zhang, Z., Gold, A., Surratt, J. D., and Thornton, J. A.: Reactive uptake of an isoprene-derived epoxydiol to submicron aerosol particles, Environ. Sci. Technol., 48, 11178–11186, https://doi.org/10.1021/es5034266, 2014.
Goliff, W. S., Stockwell, W. R., and Lawson, C. V.: The regional atmospheric chemistry mechanism, version 2, Atmos. Environ., 68, 174–185, https://doi.org/10.1016/j.atmosenv.2012.11.038, 2013.
Heinold, B., Helmert, J., Hellmuth, O., Wolke, R., Ansmann, A., Marticorena, B., Laurent, B., and Tegen, I.: Regional modeling of Saharan dust events using LM-MUSCAT: Model description and case studies, J. Geophys. Res.-Atmos., 112, D11204, https://doi.org/10.1029/2006JD007443, 2007.
Hoffmann, E. H., Tilgner, A., Schrodner, R., Brauer, P., Wolke, R., and Herrmann, H.: An advanced modeling study on the impacts and atmospheric implications of multiphase dimethyl sulfide chemistry, P. Natl. Acad. Sci. USA, 113, 11776–11781, https://doi.org/10.1073/pnas.1606320113, 2016.
Hoffmann, E. H., Tilgner, A., Vogelsberg, U., Wolke, R., and Herrmann, H.: Near-explicit multiphase modeling of halogen chemistry in a mixed urban and maritime coastal area, ACS Earth Space Chem., 3, 2452–2471, https://doi.org/10.1021/acsearthspacechem.9b00184, 2019a.
Hoffmann, E. H., Tilgner, A., Wolke, R., and Herrmann, H.: Enhanced chlorine and bromine atom activation by hydrolysis of halogen nitrates from marine aerosols at polluted coastal areas, Environ. Sci. Technol., 53, 771–778, https://doi.org/10.1021/acs.est.8b05165, 2019b.
Hossaini, R., Chipperfield, M. P., Saiz-Lopez, A., Fernandez, R., Monks, S., Feng, W. H., Brauer, P., and von Glasow, R.: A global model of tropospheric chlorine chemistry: Organic versus inorganic sources and impact on methane oxidation, J. Geophys. Res.-Atmos., 121, 14271–14297, https://doi.org/10.1002/2016jd025756, 2016.
Jacob, P. and Klockow, D.: Hydrogen peroxide measurements in the marine atmosphere, J. Atmos. Chem., 15, 353–360, https://doi.org/10.1007/BF00115404, 1992.
Jenkin, M. E., Saunders, S. M., Wagner, V., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part B): tropospheric degradation of aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 181–193, https://doi.org/10.5194/acp-3-181-2003, 2003.
Jöckel, P., Tost, H., Pozzer, A., Kunze, M., Kirner, O., Brenninkmeijer, C. A. M., Brinkop, S., Cai, D. S., Dyroff, C., Eckstein, J., Frank, F., Garny, H., Gottschaldt, K.-D., Graf, P., Grewe, V., Kerkweg, A., Kern, B., Matthes, S., Mertens, M., Meul, S., Neumaier, M., Nützel, M., Oberländer-Hayn, S., Ruhnke, R., Runde, T., Sander, R., Scharffe, D., and Zahn, A.: Earth System Chemistry integrated Modelling (ESCiMo) with the Modular Earth Submodel System (MESSy) version 2.51, Geosci. Model Dev., 9, 1153–1200, https://doi.org/10.5194/gmd-9-1153-2016, 2016.
Joshi, M., von Glasow, R., Smith, R. S., Paxton, C. G. M., Maycock, A. C., Lunt, D. J., Loptson, C., and Markwick, P.: Global warming and ocean stratification: A potential result of large extraterrestrial impacts, Geophys. Res. Lett., 44, 3841–3848, https://doi.org/10.1002/2017gl073330, 2017.
Keene, W. C. and Savoie, D. L.: Correction to “The pH of deliquesced sea-salt aerosol in polluted marine air”, Geophys. Res. Lett., 26, 1315–1316, https://doi.org/10.1029/1999gl900221, 1999.
Keene, W. C., Sander, R., Pszenny, A. A. P., Vogt, R., Crutzen, P. J., and Galloway, J. N.: Aerosol pH in the marine boundary layer: A review and model evaluation, J. Aerosol Sci., 29, 339–356, https://doi.org/10.1016/S0021-8502(97)10011-8, 1998.
Kim, Y.-M., Lee, M., Chang, W., Lee, G., Kim, K.-R., and Kato, S.: Atmospheric peroxides over the North Pacific during IOC 2002 shipboard experiment, Chemosphere, 69, 1638–1646, https://doi.org/10.1016/j.chemosphere.2007.05.057, 2007.
Kummu, M., de Moel, H., Salvucci, G., Viviroli, D., Ward, P. J., and Varis, O.: Over the hills and further away from coast: global geospatial patterns of human and environment over the 20th–21st centuries, Environ. Res. Lett., 11, 034010, https://doi.org/10.1088/1748-9326/11/3/034010, 2016.
Lana, A., Bell, T. G., Simó, R., Vallina, S. M., Ballabrera-Poy, J., Kettle, A. J., Dachs, J., Bopp, L., Saltzman, E. S., Stefels, J., Johnson, J. E., and Liss, P. S.: An updated climatology of surface dimethlysulfide concentrations and emission fluxes in the global ocean, Global Biogeochem. Cy., 25, GB1004, https://doi.org/10.1029/2010gb003850, 2011.
Law, C. S., Brévière, E., de Leeuw, G., Garçon, V., Guieu, C., Kieber, D. J., Kontradowitz, S., Paulmier, A., Quinn, P. K., Saltzman, E. S., Stefels, J., and von Glasow, R.: Evolving research directions in Surface Ocean – Lower Atmosphere (SOLAS) science, Environ. Chem., 10, 1–16, https://doi.org/10.1071/en12159, 2013.
Leser, H., Hönninger, G., and Platt, U.: MAX-DOAS measurements of BrO and NO2 in the marine boundary layer, Geophys. Res. Lett., 30, 1537, https://doi.org/10.1029/2002gl015811, 2003.
Lightfoot, P. D., Cox, R. A., Crowley, J. N., Destriau, M., Hayman, G. D., Jenkin, M. E., Moortgat, G. K., and Zabel, F.: Organic Peroxy-Radicals – Kinetics, Spectroscopy and Tropospheric Chemistry, Atmos. Environ. A, 26, 1805–1961, https://doi.org/10.1016/0960-1686(92)90423-I, 1992.
Long, M. S., Keene, W. C., Easter, R. C., Sander, R., Liu, X., Kerkweg, A., and Erickson, D.: Sensitivity of tropospheric chemical composition to halogen-radical chemistry using a fully coupled size-resolved multiphase chemistry–global climate system: halogen distributions, aerosol composition, and sensitivity of climate-relevant gases, Atmos. Chem. Phys., 14, 3397–3425, https://doi.org/10.5194/acp-14-3397-2014, 2014.
Mahajan, A. S., Oetjen, H., Lee, J. D., Saiz-Lopez, A., McFiggans, G. B., and Plane, J. M. C.: High bromine oxide concentrations in the semi-polluted boundary layer, Atmos. Environ., 43, 3811–3818, https://doi.org/10.1016/j.atmosenv.2009.05.033, 2009a.
Mahajan, A. S., Oetjen, H., Saiz-Lopez, A., Lee, J. D., McFiggans, G. B., and Plane, J. M. C.: Reactive iodine species in a semi-polluted environment, Geophys. Res. Lett., 36, L16803, https://doi.org/10.1029/2009gl038018, 2009b.
McFiggans, G., Cox, R. A., Mössinger, J. C., Allan, B. J., and Plane, J. M. C.: Active chlorine release from marine aerosols: Roles for reactive iodine and nitrogen species, J. Geophys. Res., 107, 4271, https://doi.org/10.1029/2001jd000383, 2002.
Muniz-Unamunzaga, M., Borge, R., Sarwar, G., Gantt, B., de la Paz, D., Cuevas, C. A., and Saiz-Lopez, A.: The influence of ocean halogen and sulfur emissions in the air quality of a coastal megacity: The case of Los Angeles, Sci. Total Environ., 610–611, 1536–1545, https://doi.org/10.1016/j.scitotenv.2017.06.098, 2018.
Norris, J. R.: Low cloud type over the ocean from surface observations. Part I: Relationship to surface meteorology and the vertical distribution of temperature and moisture, J. Climate, 11, 369–382, https://doi.org/10.1175/1520-0442(1998)011<0369:Lctoto>2.0.Co;2, 1998.
Pechtl, S. and von Glasow, R.: Reactive chlorine in the marine boundary layer in the outflow of polluted continental air: A model study, Geophys. Res. Lett., 34, L11813, https://doi.org/10.1029/2007gl029761, 2007.
Perraud, V., Horne, J. R., Martinez, A. S., Kalinowski, J., Meinardi, S., Dawson, M. L., Wingen, L. M., Dabdub, D., Blake, D. R., Gerber, R. B., and Finlayson-Pitts, B. J.: The future of airborne sulfur-containing particles in the absence of fossil fuel sulfur dioxide emissions, P. Natl. Acad. Sci. USA, 112, 13514–13519, https://doi.org/10.1073/pnas.1510743112, 2015.
Pszenny, A. A. P., Moldanová, J., Keene, W. C., Sander, R., Maben, J. R., Martinez, M., Crutzen, P. J., Perner, D., and Prinn, R. G.: Halogen cycling and aerosol pH in the Hawaiian marine boundary layer, Atmos. Chem. Phys., 4, 147–168, https://doi.org/10.5194/acp-4-147-2004, 2004.
Quinn, P. K., Collins, D. B., Grassian, V. H., Prather, K. A., and Bates, T. S.: Chemistry and related properties of freshly emitted sea spray aerosol, Chem. Rev., 115, 4383–4399, https://doi.org/10.1021/cr500713g, 2015.
Read, K. A., Mahajan, A. S., Carpenter, L. J., Evans, M. J., Faria, B. V., Heard, D. E., Hopkins, J. R., Lee, J. D., Moller, S. J., Lewis, A. C., Mendes, L., McQuaid, J. B., Oetjen, H., Saiz-Lopez, A., Pilling, M. J., and Plane, J. M.: Extensive halogen-mediated ozone destruction over the tropical Atlantic Ocean, Nature, 453, 1232–1235, https://doi.org/10.1038/nature07035, 2008.
Saiz-Lopez, A. and von Glasow, R.: Reactive halogen chemistry in the troposphere, Chem. Soc. Rev., 41, 6448–6472, https://doi.org/10.1039/c2cs35208g, 2012.
Saiz-Lopez, A., Plane, J. M., Baker, A. R., Carpenter, L. J., von Glasow, R., Martin, J. C., McFiggans, G., and Saunders, R. W.: Atmospheric chemistry of iodine, Chem. Rev., 112, 1773–1804, https://doi.org/10.1021/cr200029u, 2012.
Saiz-Lopez, A., Fernandez, R. P., Ordóñez, C., Kinnison, D. E., Gómez Martín, J. C., Lamarque, J.-F., and Tilmes, S.: Iodine chemistry in the troposphere and its effect on ozone, Atmos. Chem. Phys., 14, 13119–13143, https://doi.org/10.5194/acp-14-13119-2014, 2014.
Sander, R., Keene, W. C., Pszenny, A. A. P., Arimoto, R., Ayers, G. P., Baboukas, E., Cainey, J. M., Crutzen, P. J., Duce, R. A., Hönninger, G., Huebert, B. J., Maenhaut, W., Mihalopoulos, N., Turekian, V. C., and Van Dingenen, R.: Inorganic bromine in the marine boundary layer: a critical review, Atmos. Chem. Phys., 3, 1301–1336, https://doi.org/10.5194/acp-3-1301-2003, 2003.
Sander, R., Pszenny, A. A. P., Keene, W. C., Crete, E., Deegan, B., Long, M. S., Maben, J. R., and Young, A. H.: Gas phase acid, ammonia and aerosol ionic and trace element concentrations at Cape Verde during the Reactive Halogens in the Marine Boundary Layer (RHaMBLe) 2007 intensive sampling period, Earth Syst. Sci. Data, 5, 385–392, https://doi.org/10.5194/essd-5-385-2013, 2013.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Schmidt, J. A., Jacob, D. J., Horowitz, H. M., Hu, L., Sherwen, T., Evans, M. J., Liang, Q., Suleiman, R. M., Oram, D. E., Le Breton, M., Percival, C. J., Wang, S., Dix, B., and Volkamer, R.: Modeling the observed tropospheric BrO background: Importance of multiphase chemistry and implications for ozone, OH, and mercury, J. Geophys. Res.-Atmos, 121, 11819–811835, https://doi.org/10.1002/2015jd024229, 2016.
Schrödner, R., Tilgner, A., Wolke, R., and Herrmann, H.: Modeling the multiphase processing of an urban and a rural air mass with COSMO–MUSCAT, Urban Clim., 10, 720–731, https://doi.org/10.1016/j.uclim.2014.02.001, 2014.
Schrödner, R., Wolke, R., Tilgner, A., van Pinxteren, D., and Herrmann, H.: Modelling Multiphase Aerosol-Cloud Processing with the 3-D CTM COSMO-MUSCAT: Application for Cloud Events During HCCT-2010, Air Pollution Modeling and its Application XXV, Cham, 587–592, 2018.
Schultz, M. G., Stadtler, S., Schröder, S., Taraborrelli, D., Franco, B., Krefting, J., Henrot, A., Ferrachat, S., Lohmann, U., Neubauer, D., Siegenthaler-Le Drian, C., Wahl, S., Kokkola, H., Kühn, T., Rast, S., Schmidt, H., Stier, P., Kinnison, D., Tyndall, G. S., Orlando, J. J., and Wespes, C.: The chemistry–climate model ECHAM6.3-HAM2.3-MOZ1.0, Geosci. Model Dev., 11, 1695–1723, https://doi.org/10.5194/gmd-11-1695-2018, 2018.
Sehili, A. M., Wolke, R., Knoth, O., Simmel, M., Tilgner, A., and Herrmann, H.: Comparison of different model approaches for the simulation of multiphase processes, Atmos. Environ., 39, 4403–4417, https://doi.org/10.1016/j.atmosenv.2005.02.039, 2005.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric Chemistry and Physics, John Wiley & Sons, Inc., Hoboken, New Jersey, USA, 1248 pp., 2006.
Shechner, M. and Tas, E.: Ozone Formation Induced by the Impact of Reactive Bromine and Iodine Species on Photochemistry in a Polluted Marine Environment, Environ. Sci. Technol., 51, 14030–14037, https://doi.org/10.1021/acs.est.7b02860, 2017.
Sherwen, T., Schmidt, J. A., Evans, M. J., Carpenter, L. J., Großmann, K., Eastham, S. D., Jacob, D. J., Dix, B., Koenig, T. K., Sinreich, R., Ortega, I., Volkamer, R., Saiz-Lopez, A., Prados-Roman, C., Mahajan, A. S., and Ordóñez, C.: Global impacts of tropospheric halogens (Cl, Br, I) on oxidants and composition in GEOS-Chem, Atmos. Chem. Phys., 16, 12239–12271, https://doi.org/10.5194/acp-16-12239-2016, 2016.
Sherwen, T., Evans, M. J., Sommariva, R., Hollis, L. D. J., Ball, S. M., Monks, P. S., Reed, C., Carpenter, L. J., Lee, J. D., Forster, G., Bandy, B., Reeves, C. E., and Bloss, W. J.: Effects of halogens on European air-quality, Faraday Discuss., 200, 75–100, https://doi.org/10.1039/c7fd00026j, 2017.
Simpson, W. R., Brown, S. S., Saiz-Lopez, A., Thornton, J. A., and Glasow, R.: Tropospheric halogen chemistry: sources, cycling, and impacts, Chem. Rev., 115, 4035–4062, https://doi.org/10.1021/cr5006638, 2015.
Steppeler, J., Doms, G., Schattler, U., Bitzer, H. W., Gassmann, A., Damrath, U., and Gregoric, G.: Meso-gamma scale forecasts using the nonhydrostatic model LM, Meteorol. Atmos. Phys., 82, 75–96, https://doi.org/10.1007/s00703-001-0592-9, 2003.
Surratt, J. D., Lewandowski, M., Offenberg, J. H., Jaoui, M., Kleindienst, T. E., Edney, E. O., and Seinfeld, J. H.: Effect of Acidity on Secondary Organic Aerosol Formation from Isoprene, Environ. Sci. Technol., 41, 5363–5369, https://doi.org/10.1021/es0704176, 2007.
Surratt, J. D., Chan, A. W., Eddingsaas, N. C., Chan, M., Loza, C. L., Kwan, A. J., Hersey, S. P., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Reactive intermediates revealed in secondary organic aerosol formation from isoprene, P. Natl. Acad. Sci. USA, 107, 6640–6645, https://doi.org/10.1073/pnas.0911114107, 2010.
Volkamer, R., Baidar, S., Campos, T. L., Coburn, S., DiGangi, J. P., Dix, B., Eloranta, E. W., Koenig, T. K., Morley, B., Ortega, I., Pierce, B. R., Reeves, M., Sinreich, R., Wang, S., Zondlo, M. A., and Romashkin, P. A.: Aircraft measurements of BrO, IO, glyoxal, NO2, H2O, O2–O2 and aerosol extinction profiles in the tropics: comparison with aircraft-/ship-based in situ and lidar measurements, Atmos. Meas. Tech., 8, 2121–2148, https://doi.org/10.5194/amt-8-2121-2015, 2015.
von Glasow, R. and Crutzen, P. J.: Model study of multiphase DMS oxidation with a focus on halogens, Atmos. Chem. Phys., 4, 589–608, https://doi.org/10.5194/acp-4-589-2004, 2004.
von Glasow, R., Sander, R., Bott, A., and Crutzen, P. J.: Modeling halogen chemistry in the marine boundary layer 2. Interactions with sulfur and the cloud-covered MBL, J. Geophys. Res.-Atmos., 107, 4323, https://doi.org/10.1029/2001jd000943, 2002a.
von Glasow, R., Sander, R., Bott, A., and Crutzen, P. J.: Modeling halogen chemistry in the marine boundary layer 1. Cloud-free MBL, J. Geophys. Res.-Atmos., 107, 4341, https://doi.org/10.1029/2001jd000942, 2002b.
von Glasow, R., Jickells, T. D., Baklanov, A., Carmichael, G. R., Church, T. M., Gallardo, L., Hughes, C., Kanakidou, M., Liss, P. S., Mee, L., Raine, R., Ramachandran, P., Ramesh, R., Sundseth, K., Tsunogai, U., Uematsu, M., and Zhu, T.: Megacities and large urban agglomerations in the coastal zone: interactions between atmosphere, land, and marine ecosystems, Ambio, 42, 13–28, https://doi.org/10.1007/s13280-012-0343-9, 2013.
Wang, X., Jacob, D. J., Eastham, S. D., Sulprizio, M. P., Zhu, L., Chen, Q., Alexander, B., Sherwen, T., Evans, M. J., Lee, B. H., Haskins, J. D., Lopez-Hilfiker, F. D., Thornton, J. A., Huey, G. L., and Liao, H.: The role of chlorine in global tropospheric chemistry, Atmos. Chem. Phys., 19, 3981–4003, https://doi.org/10.5194/acp-19-3981-2019, 2019.
Wolke, R., Knoth, O., Hellmuth, O., Schröder, W., and Renner, E.: The parallel model system LM-MUSCAT for chemistry-transport simulations: Coupling scheme, parallelization and application, in: Parallel Computing: Software Technology, Algorithms, Architectures, and Applications, edited by: Joubert, G. R., Nagel, W. E., Peters, F. J., and Walter, W. V., Elsevier, Amsterdam, Netherlands, 363–370, 2004.
Wolke, R., Sehili, A. M., Simmel, M., Knoth, O., Tilgner, A., and Herrmann, H.: SPACCIM: A parcel model with detailed microphysics and complex multiphase chemistry, Atmos. Environ., 39, 4375–4388, https://doi.org/10.1016/j.atmosenv.2005.02.038, 2005.
Wolke, R., Schröder, W., Schrödner, R., and Renner, E.: Influence of grid resolution and meteorological forcing on simulated European air quality: A sensitivity study with the modeling system COSMO–MUSCAT, Atmos. Environ., 53, 110–130, https://doi.org/10.1016/j.atmosenv.2012.02.085, 2012.
Wu, R., Wang, S., and Wang, L.: New mechanism for the atmospheric oxidation of dimethyl sulfide. The importance of intramolecular hydrogen shift in a CH3SCH2OO radical, J. Phys. Chem. A, 119, 112–117, https://doi.org/10.1021/jp511616j, 2015.
Wudl, F., Lightner, D. A., and Cram, D. J.: Methanesulfinic acid and Its properties, J. Am. Chem. Soc., 89, 4099–4101, https://doi.org/10.1021/Ja00992a026, 1967.
Xue, L. K., Saunders, S. M., Wang, T., Gao, R., Wang, X. F., Zhang, Q. Z., and Wang, W. X.: Development of a chlorine chemistry module for the Master Chemical Mechanism, Geosci. Model Dev., 8, 3151–3162, https://doi.org/10.5194/gmd-8-3151-2015, 2015.
Yan, Y., Cabrera-Perez, D., Lin, J., Pozzer, A., Hu, L., Millet, D. B., Porter, W. C., and Lelieveld, J.: Global tropospheric effects of aromatic chemistry with the SAPRC-11 mechanism implemented in GEOS-Chem version 9-02, Geosci. Model Dev., 12, 111–130, https://doi.org/10.5194/gmd-12-111-2019, 2019.
Yin, F. D., Grosjean, D., and Seinfeld, J. H.: Photooxidation of dimethyl sulfide and dimethyl disulfide. 1. Mechanism Development, J. Atmos. Chem., 11, 309–364, https://doi.org/10.1007/Bf00053780, 1990.
Yuan, J. and Shiller, A. M.: The variation of hydrogen peroxide in rainwater over the South and Central Atlantic Ocean, Atmos. Environ., 34, 3973–3980, https://doi.org/10.1016/S1352-2310(00)00167-9, 2000.
Zängl, G., Reinert, D., Rípodas, P., and Baldauf, M.: The ICON (ICOsahedral Non-hydrostatic) modelling framework of DWD and MPI-M: Description of the non-hydrostatic dynamical core, Q. J. Roy. Meteorol. Soc., 141, 563–579, https://doi.org/10.1002/qj.2378, 2015.
Zhu, L., Nicovich, J. M., and Wine, P. H.: Temperature-dependent kinetics studies of aqueous phase reactions of hydroxyl radicals with dimethylsulfoxide, dimethylsulfone, and methanesulfonate, Aquat. Sci., 65, 425–435, https://doi.org/10.1007/s00027-003-0673-6, 2003.
Zhu, L., Jacob, D. J., Eastham, S. D., Sulprizio, M. P., Wang, X., Sherwen, T., Evans, M. J., Chen, Q., Alexander, B., Koenig, T. K., Volkamer, R., Huey, L. G., Le Breton, M., Bannan, T. J., and Percival, C. J.: Effect of sea salt aerosol on tropospheric bromine chemistry, Atmos. Chem. Phys., 19, 6497–6507, https://doi.org/10.5194/acp-19-6497-2019, 2019.