the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 01 Feb 2019
| 01 Feb 2019
Global aerosol modeling with MADE3 (v3.0) in EMAC (based on v2.53): model description and evaluation
J. Christopher Kaiser
Johannes Hendricks
Patrick Jöckel
Holger Tost
Konrad Kandler
Bernadett Weinzierl
Daniel Sauer
Katharina Heimerl
Joshua P. Schwarz
Anne E. Perring
Thomas Popp
Recently, the aerosol microphysics submodel MADE3 (Modal Aerosol Dynamics model for Europe, adapted for global applications, third generation) was introduced as a successor to MADE and MADE-in. It includes nine aerosol species and nine lognormal modes to represent aerosol particles of three different mixing states throughout the aerosol size spectrum. Here, we describe the implementation of the most recent version of MADE3 into the ECHAM/MESSy Atmospheric Chemistry (EMAC) general circulation model, including a detailed evaluation of a 10-year aerosol simulation with MADE3 as part of EMAC.
We compare simulation output to station network measurements of near-surface aerosol component mass concentrations, to airborne measurements of aerosol mass mixing ratio and number concentration vertical profiles, to ground-based and airborne measurements of particle size distributions, and to station network and satellite measurements of aerosol optical depth. Furthermore, we describe and apply a new evaluation method, which allows a comparison of model output to size-resolved electron microscopy measurements of particle composition. Although there are indications that fine-mode particle deposition may be underestimated by the model, we obtained satisfactory agreement with the observations. Remaining deviations are of similar size to those identified in other global aerosol model studies.
Thus, MADE3 can be considered ready for application within EMAC. Due to its detailed representation of aerosol mixing state, it is especially useful for simulating wet and dry removal of aerosol particles, aerosol-induced formation of cloud droplets and ice crystals as well as aerosol–radiation interactions. Besides studies on these fundamental processes, we also plan to use MADE3 for a reassessment of the climate effects of anthropogenic aerosol perturbations.
- Article
(3887 KB) - Full-text XML
-
Supplement
(222 KB) - BibTeX
- EndNote





The MESSy (Modular Earth Submodel System; Jöckel et al., 2010) aerosol microphysics submodel MADE3 (Modal Aerosol Dynamics model for Europe, adapted for global applications, third generation) was created with a requantification of the aerosol–climate effect of offshore ship traffic in mind (Kaiser et al., 2014). Simulations in the past indicated that sulfate formed from ship emissions may be one of the major contributors to the negative anthropogenic aerosol radiative forcing (Capaldo et al., 1999; Lauer et al., 2007, 2009; Righi et al., 2011, 2013; Olivié et al., 2012; Peters et al., 2012, 2013). Due to increasingly stringent regulations on the maximum sulfur content of ship fuels (IMO, 2011), this contribution is expected to decrease strongly in the future (Lauer et al., 2009; Righi et al., 2011; Schembari et al., 2012; Johansson et al., 2013; Jonson et al., 2015). Possibly, aerosol nitrate formation will compensate for part of the reduction (Lauer et al., 2009; Bellouin et al., 2011; Righi et al., 2011). Measurements indicated that it may be crucial to take into account coarse-mode particle interactions with condensable trace gases in order to quantify this effect (Kerminen et al., 1997; Hara et al., 1999; Yeatman et al., 2001; Cavalli et al., 2004; Nolte et al., 2008; Prabhakar et al., 2014). Such interactions were neglected in the previous assessments and were therefore included in MADE3, which represents an extension of MADE (Lauer et al., 2007) and its successor MADE-in with enhanced resolution of fine particle mixing state (Aquila et al., 2011).
As an improvement to its predecessors, MADE3 includes computationally efficient and consistent representations of three different aerosol mixing states in each of three different size ranges, which can be advantageous for many other applications. For instance, we aim to use MADE3 for assessments of aerosol–ice cloud interactions. Particles composed of compounds with no or very low water solubility (in the following denoted as “insoluble particles”), such as mineral dust or black carbon particles, can serve as ice nuclei initiating ice formation in mixed-phase or cirrus clouds (e.g., Lohmann and Feichter, 2005; Hoose and Möhler, 2012). The ice formation efficiency of these particles strongly depends on their size, surface area, and state of mixing with soluble aerosol species. To simulate these effects, climate models should allow for explicit predictions of the number concentration, size distribution, and mixing state of aerosol particles containing insoluble components. In the first generation of model studies on the role of ice nuclei in the global climate system, bulk aerosol schemes were applied (Lohmann et al., 2004; Hendricks et al., 2005, 2011). This implied that the number of potential ice nuclei had to be estimated from aerosol mass assuming typical aerosol size distributions. Advanced aerosol schemes allowing the explicit simulation of the aerosol number concentration and size distribution were applied in more recent studies (e.g., Lohmann and Hoose, 2009; Gettelman et al., 2012; Kuebbeler et al., 2014; Zhou and Penner, 2014). However, the individual number concentrations of insoluble particles in different size ranges and mixing states could only partly be quantified with these approaches. MADE3 has the advantage that it allows explicit simulations of the number concentration, size distribution (assuming lognormal modes with fixed widths), and mixing state (external or internal mixture) of aerosol particles containing insoluble components. Hence, the new aerosol scheme opens new opportunities for the simulation of aerosol effects on ice clouds.
We intend to use the enhanced capabilities of MADE3 to update the results of our previous studies on the health and climate impacts of the transport sectors (Corbett et al., 2007; Lauer et al., 2009; Winebrake et al., 2009; Righi et al., 2011, 2013, 2015, 2016; Lund et al., 2012). Such model applications will be the subject of future investigations. As a first step towards these studies, the present article provides a detailed description of the implementation of MADE3 into a global chemistry–climate model as well as an evaluation of a first reference simulation.
The ability of the MADE3 algorithms to solve the gas–aerosol partitioning (outside clouds), new particle formation, and coagulation parts of the aerosol dynamics equation was demonstrated by Kaiser et al. (2014). For the solution of the full equation, a number of further processes have to be considered, namely particle and precursor emissions; particle transport by advection, convection, and turbulent diffusion; aerosol precursor chemistry in the gas and liquid phases; and cloud and precipitation scavenging of aerosols, as well as their dry deposition and sedimentation. Hence, we describe here the implementation of MADE3 into the atmospheric chemistry general circulation model EMAC (ECHAM/MESSy Atmospheric Chemistry; Jöckel et al., 2010, 2016), which includes further submodels to represent these processes (Sect. 2). Subsequently, in Sect. 3, we present an evaluation of the performance of EMAC with MADE3 as a global aerosol model. The evaluation is accomplished by comparison of EMAC aerosol simulations to observational data from a multitude of different sources, including station networks, airborne measurements, laboratory analyses of in situ sampled particles, and satellite data. The main conclusions of this study are summarized in Sect. 4. Appendix A provides a list of the acronyms used in this article. Details of the aerosol scavenging scheme are explained in Appendix B. Descriptions of the chemistry mechanisms considered are provided in the Supplement.
The work presented in this paper is partly based on the PhD thesis by J. C. Kaiser (Kaiser, 2016). We therefore explain to the reader that significant parts of the text in the abstract, Sects. 2 and 3, and Appendix B already appeared in Kaiser (2016).
2.1 EMAC setup
The EMAC model is a numerical chemistry and climate simulation system that includes submodels describing tropospheric and middle atmospheric processes and their interaction with oceans, land, and human influences (Jöckel et al., 2010). It uses the second version of MESSy to link multi-institutional computer codes. The core atmospheric model is the ECHAM5 (fifth-generation European Centre Hamburg) general circulation model (Roeckner et al., 2006). For the present study, we applied EMAC (ECHAM5 version 5.3.02, MESSy version 2.53) in the T42L19 resolution, i.e., with a spherical truncation of T42 (corresponding to a quadratic Gaussian grid of approximately 2.8 by 2.8 degrees in latitude and longitude) with 19 vertical hybrid sigma-pressure levels up to 10 hPa. The applied model setup comprised the submodels given in Table 1. A model time step length Δt of 30 min was used and a temporal resolution for the simulation output of 12 h.
Dietmüller et al. (2016)Roeckner et al. (2006)Dietmüller et al. (2016)Tost et al. (2006b)Tost et al. (2010)Kerkweg et al. (2006a, 2009)Jöckel et al. (2006)Sander et al. (2014)Tost et al. (2007)Kaiser et al. (2014)Sander et al. (2011)Kerkweg et al. (2006b)Kerkweg et al. (2006b)Dietmüller et al. (2016)Dietmüller et al. (2016)Tost et al. (2006a, 2010)Kerkweg et al. (2006a)Jöckel et al. (2016)Kerkweg et al. (2006b)Jöckel et al. (2006)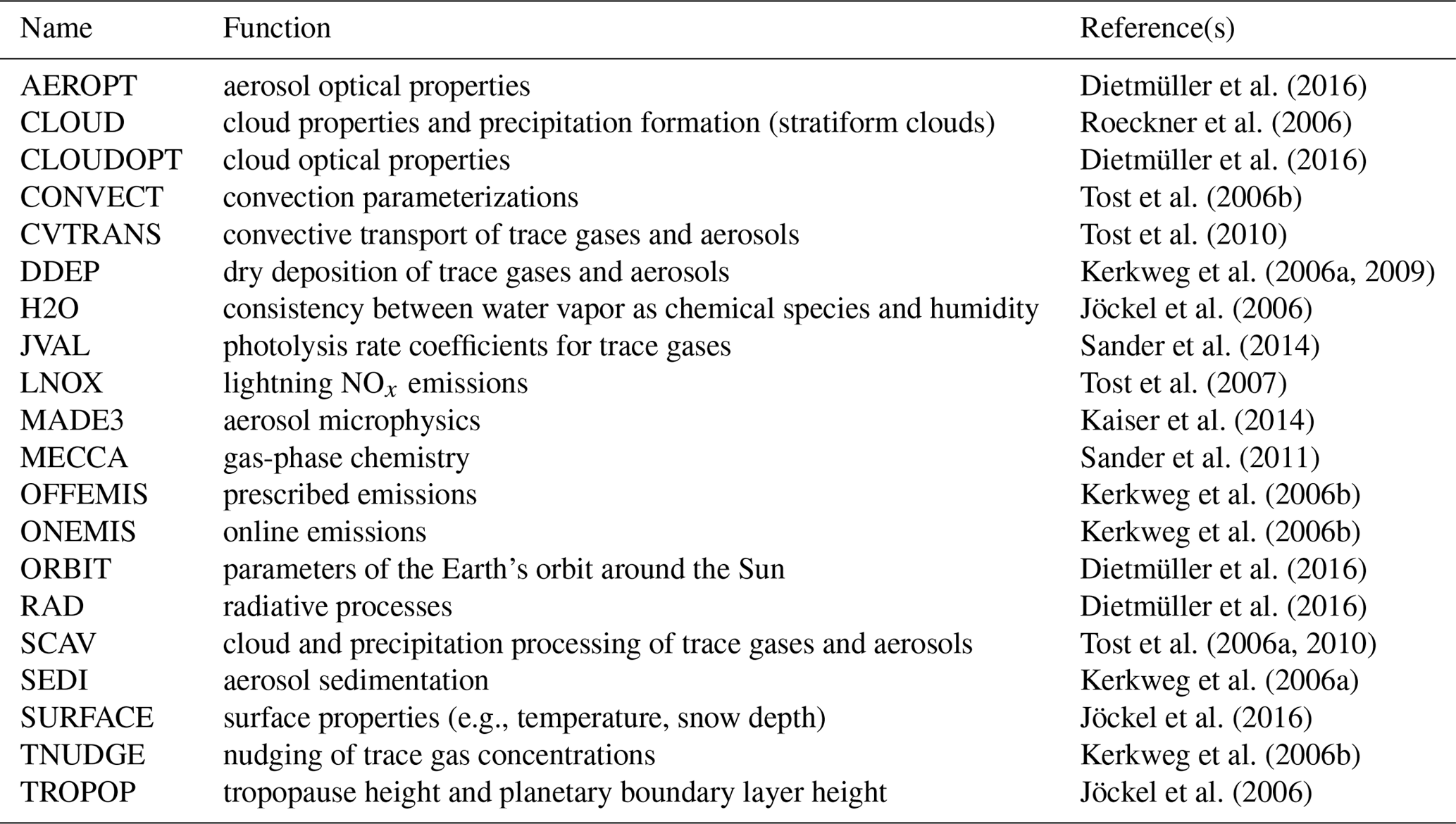
We simulated 11 years in “nudged mode”; i.e., wind divergence and vorticity, temperature, and logarithm of the surface pressure were relaxed towards ERA-Interim reanalyses for the years 1995–2005. The first simulated year is regarded as the (aerosol) spin-up phase, so that our evaluation only takes into account the 10 years that followed.
Emissions of both gases and aerosol particles enter the EMAC atmosphere through the submodels OFFEMIS, for prescribed emissions, and ONEMIS, for so-called online emissions that depend on the dynamics of the atmosphere (e.g., wind speed) and the state of its lower boundary (e.g., sea surface temperature). The emission setup used here is described in a separate subsection (Sect. 2.4). Prescribed emissions are representative of the year 2000. The production of nitrogen oxides (NOx, i.e., NO and NO2) from lightning was taken into account by the submodel LNOX, using a parameterization by Price and Rind (1992), which is based on convective cloud top height as the driving parameter. The parameterization was tuned to match global total emissions within the observed range (Schumann and Huntrieser, 2007).
Aerosol particle transport is part of the tracer advection and vertical diffusion schemes of the base model ECHAM5 and of the convective transport submodel CVTRANS. Horizontal diffusion of particles is not considered in ECHAM5, but it is anyway not expected to contribute significantly to transport on the scales of the model grid boxes as used here.
We used the submodel MECCA to simulate atmospheric gas-phase chemistry. For computational efficiency, the simplified tropospheric chemistry scheme that was created by Lauer et al. (2007) was used. It includes 34 gases and 60 chemical reactions (47 gas-phase and 13 photolysis reactions) to describe chemistry and the tropospheric sulfur cycle (see the Supplement for more details). The photolysis rates are calculated by the submodel JVAL. Heterogeneous reactions, i.e., reactions of trace gases on or with aerosol particle surfaces, are not included. Note, however, that reactions on cloud droplet surfaces are included via the cloud-phase chemistry (see Sect. 2.3 and the Supplement).
MADE3, which is used for the representation of aerosol microphysics, will be described in Sect. 2.2. To avoid convoluting the results with feedbacks from the simulated aerosol on model dynamics, we switch off the feedback of the MADE3 aerosol on clouds and radiation in the model configuration described here. Before such feedbacks will be considered, the quality of the MADE3 aerosol has to be proven, which is the purpose of this study.
Deposition of aerosol particles is handled in EMAC by the submodels DDEP, which uses the so-called “big leaf” approach assuming that deposition fluxes within the canopy have the same relative responses to the environment as any single leaf, and that the scaling from leaf to canopy is therefore linear (Sellers et al., 1996); SEDI, for sedimentation (gravitational settling); and SCAV, for wet deposition. The latter required some MADE3-specific modifications; see Sect. 2.3.
Optical properties of aerosol particles, which are considered to compute aerosol optical depth (AOD) for comparison with satellite data (Sect. 3.5), are determined by the submodel AEROPT. The lookup tables applied in AEROPT are the same for MADE3 and its predecessor MADE. Hence, we used the MADE tables that were created by Lauer et al. (2007) with the help of the software libRadtran (Mayer and Kylling, 2005). Further details on these calculations are provided by Dietmüller et al. (2016).
Cloud properties are calculated by the submodels CLOUD (stratiform clouds) and CONVECT (convective clouds) in EMAC. For stratiform clouds, we selected the standard ECHAM5 single-moment cloud scheme (Roeckner et al., 2003), i.e., a scheme that only considers water and ice mass but no droplet or ice crystal numbers. Although previous studies with the MADE3 predecessors were carried out with two-moment cloud schemes, a single-moment scheme is sufficient here, as we do not attempt to quantify the climatic impact of aerosol particles. This will be the subject of follow-up studies, however. Instead of the Tompkins (2002) method to calculate fractional cloud cover (as described by Roeckner et al., 2003), we choose the parameterization developed by Sundqvist et al. (1989). The high numerical stability of this scheme is advantageous for multi-year climate simulations. For convective clouds, we choose the original ECHAM5 scheme (Roeckner et al., 2003), which is based on work by Tiedtke (1989) and Nordeng (1994), with modifications by Brinkop and Sausen (1997).
2.2 MADE3 v3.0
MADE3 was described in detail by Kaiser et al. (2014). Therefore, we only briefly repeat its main characteristics here and in Fig. 1. The aerosol is represented by the modal approach, namely with nine modes that represent different particle mixing states and different particle size ranges. Each of the Aitken, accumulation, and coarse-mode size ranges in MADE3 includes three modes: one for particles fully composed of water-soluble species, one for particles mainly composed of insoluble material (i.e., insoluble particles with only very thin coatings of soluble material), and one for mixed particles (i.e., particles composed of soluble material including insoluble immersions). In the following, we will refer to these modes as “soluble”, “insoluble”, and “mixed” modes, respectively. The considered components that make up these modes are sulfate (SO4), ammonium (NH4), nitrate (NO3), sea spray (SS) components other than chloride (mainly sodium; Na), chloride (Cl), particulate organic matter (POM), black carbon (BC), mineral dust (DU), and aerosol water (H2O). Different from the MADE3 box model version presented by Kaiser et al. (2014), where the insoluble modes were dominated by BC and mineral dust, we now also assign hydrophobic POM to the insoluble modes during emission (see below) in order to describe interactions of this aerosol component with clouds more consistently. Observations also show that BC is mostly emitted internally mixed with POM (e.g., Petzold et al., 2013).
MADE3 calculates changes in the particle number concentration, size distribution, and composition due to gas–particle partitioning, particle coagulation, and new particle formation. For the gas–particle partitioning of semi-volatile species, i.e., ammonia (NH3)∕NH4, nitric acid (HNO3)∕NO3, and hydrochloric acid (HCl)∕Cl, an equilibrium approach is applied, where condensation towards the coarse-mode particles is limited to the kinetically possible fluxes. Sulfuric acid (H2SO4) and secondary organic aerosol (SOA) precursors are assumed to condense irreversibly on the particles. The amount of condensable H2SO4 is calculated online by the model using the corresponding production rate as provided by the chemical scheme. The amount of condensable SOA is prescribed in terms of an effective emission of SOA from natural terpenes based on Dentener et al. (2006). The aerosol dynamics equation is solved by applying a combination of analytical approximations and process-specific numerical solvers. For the details of this approach, we refer to Kaiser et al. (2014) and references therein. In addition to some technical changes between the MADE3 version described by Kaiser et al. (2014) (v2.0b) and the one used here (v3.0), we modified the treatment of new particles upon nucleation events, as well as the criterion for transferring particles from the insoluble to the mixed modes, denoted as the aging criterion, as follows.
In the original version of the model, the transfer to the mixed modes was induced as soon as insoluble particles obtained a liquid coating of a critical size. We now neglect aerosol water in this aging calculation and, correspondingly, in the target mode assignment upon particle coagulation. Hence, only the water-soluble components of the coating are taken into account. In this way, we interpret water uptake as a consequence of particle aging rather than as the cause of it. We further neglect the POM fraction in these model operations since its role in the aging process is still uncertain. Particles from the insoluble modes are now transferred to the mixed modes if the sum of the soluble inorganic component masses exceeds 10 % of the modal dry mass. This assumption is supported by laboratory and field measurements as reported by Svenningsson et al. (1994), Khalizov et al. (2009), and Liu et al. (2013). Correspondingly, we assign particles that result from coagulation of insoluble modes with mixed or soluble modes to an insoluble mode when the resulting soluble inorganic contribution to dry mass is less than 10 %, and to a mixed mode otherwise.
Concerning nucleation, we now account for initial growth of particles at unresolved temporal and spatial scales by rescaling the formation rate of H2SO4–H2O particles with a mode median wet diameter of 3.5 nm (as it was formerly implemented) to a formation rate of SO4 particles with a mode median dry diameter of 10 nm. This basically corresponds to redistributing the nucleated mass into larger particles, assuming a lognormal size distribution with the same width but with a larger median diameter, which results in a decrease in nucleated particle number. With this modification, the model seems to perform better at accurately simulating particle evolution, as evidenced by the comparisons of number concentrations and size distributions to observations in the free troposphere, where nucleation is the major source of ultrafine particles (see Sect. 3.2 and 3.3). We base this updated assumption on new particle formation measurements as reported by, e.g., Modini et al. (2009), Kerminen et al. (2010), Boulon et al. (2011), Matsui et al. (2011), Young et al. (2013), García et al. (2014), Chandra et al. (2016), Giamarelou et al. (2016), and Ueda et al. (2016).
When coupling the MADE3 aerosol to the cloud processing submodel SCAV (see Appendix B), we assume that the insoluble modes are hydrophobic, whereas we consider mixed and soluble modes to be hydrophilic. Hence, only particles in the mixed and soluble modes undergo liquid nucleation scavenging; i.e., they can serve as nuclei for cloud droplet formation. In contrast, ice nucleation scavenging is considered to be less efficient for purely soluble particles (see Appendix B1). Analogous to Aquila et al. (2011), we assume that 5 % of the soluble particles are incorporated into ice crystals (ice nucleation scavenging ratio of 0.05) in cirrus clouds (T≤238.15 K), consistent with scavenging ratios typical for homogeneous freezing of aerosols. In order to account for the ability of insoluble particles to act as ice nuclei in heterogeneous ice formation processes, we assume a higher scavenging ratio, namely 0.1, for particles containing BC or dust, either externally or internally mixed (insoluble and mixed modes). In the mixed-phase cloud regime (T>238.15 K), we assume an ice nucleation scavenging ratio of 0.1 for all particle types. This rough estimate is based on the fact that, due to the limited number of ice nuclei, only a fraction of cloud droplets freezes during glaciation of liquid clouds, while the majority of the droplets evaporate via the Bergeron–Findeisen process, thereby releasing large amounts of aerosol mass originally scavenged during liquid droplet formation. A ratio of 0.1 corresponds to typical ratios of the concentrations of ice crystals and cloud droplets in this regime (Korolev et al., 2003). Impaction scavenging does not depend on the particle type.
Due to the extended mode structure of MADE3 with respect to the other two aerosol submodels that can be used with SCAV in EMAC, i.e., the first version of MADE and the Global Modal-aerosol eXtension (GMXe) submodel (Pringle et al., 2010), a number of modifications to SCAV were required. The main conceptual difference is described in the following subsection.
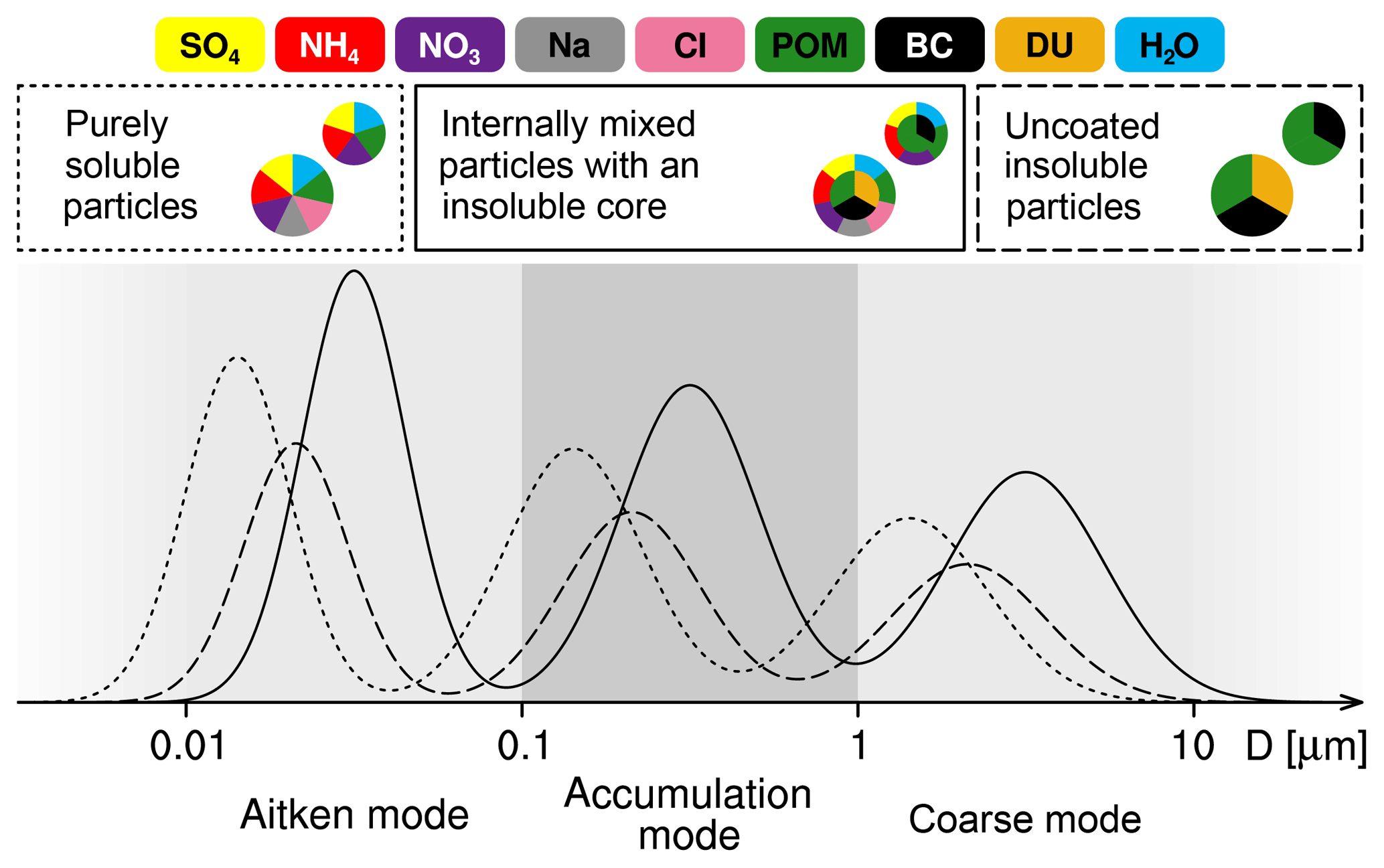
Figure 1Schematic representation of the MADE3 submodel. The colors represent the different chemical components. The dotted, solid, and dashed lines correspond to the different mixing states (soluble, mixed, and insoluble, respectively).
2.3 Aerosol processing in clouds and precipitation
Cloud and precipitation chemistry as well as wet deposition of both aerosol and gas species are treated by the submodel SCAV in EMAC (see Appendix B). We neglect ice-phase chemistry here (including the uptake of gases onto ice particles) but include 35 chemical components and 45 reactions in the liquid-phase chemistry scheme. Heterogeneous processes like the formation of aqueous HNO3 from gaseous N2O5 on droplet surfaces are also considered. We refer the reader to the Supplement for more details on the chemical mechanisms adopted in this work. For a description of the model representation of the different aerosol scavenging processes, we refer to Appendix B1.
Resuspension of aerosol particles from evaporating/sublimating cloud particles or precipitating hydrometeors is an important process to be represented, which has recently been confirmed by Gao et al. (2016). For MADE3, we completely revised the routines describing the redistribution of the resuspended aerosol. The basic assumptions for the assignment of such residual aerosol to the MADE3 modes are described below. Technical details and mathematics are provided in Appendix B2. Note that, in the following, (i) “cloud particles” refers to both ice crystals and liquid droplets suspended in clouds, as well as to falling snowflakes and rain droplets; (ii) “cloud residual aerosol” also includes “precipitation residual aerosol”; and (iii) “evaporation” includes both evaporation of cloud and rain droplets, and sublimation or melting plus subsequent evaporation of ice crystals and snowflakes. The following assumptions are made:
-
According to an operator splitting approach, we assume that activation of aerosol particles into cloud particles occurs first, and impaction of interstitial aerosol upon the cloud particles follows in an instantaneous manner as a second step. We acknowledge that this constitutes a strong simplification of the two interdependent processes, which may influence the simulation of the cloud residual aerosol. When more measurement data on the mixing state of cloud residual aerosol become available, the impact of this simplification can be evaluated and the algorithm may then be refined. However, as the influence of cloud particle coagulation on mixing state is not represented in the model code, there will be some inevitable error anyway.
-
In order to keep the complexity of the involved equations at a reasonable level and to avoid underestimations of aerosol transformations within clouds, we further assume that, during impaction scavenging, the interactions of interstitial aerosol with cloud particles are as homogeneously distributed as possible over the cloud particle population, regardless of the cloud particles' aerosol cores (see Appendix B2 for more details on this assumption).
-
Aerosol particles from the insoluble modes (dashed line in Fig. 1) cannot be activated into cloud droplets in the present scheme, as they are assumed to be hydrophobic. Nevertheless, they can serve as ice nuclei.
-
The chemical formation of water-soluble species within cloud droplets and coagulation between cloud particles lead to accumulation of soluble aerosol components inside cloud particles. To account for such effects, we assume that all aerosol particles that were incorporated into cloud particles are hydrophilic upon cloud particle evaporation. Hence, no residual aerosol is assigned to the insoluble, hydrophobic modes.
-
Furthermore, we assume that – due to collection of other aerosol particles, generation of aerosol mass inside cloud droplets, and coagulation of cloud particles – aerosol particle cores of the cloud particles resulting from activated Aitken-mode aerosol will have grown from Aitken- to accumulation- or coarse-mode sizes when the cloud particles evaporate. Hence, no residual aerosol is assigned to the Aitken modes.
2.4 Emissions setup
The emissions setup for the present study was in large parts designed by Righi et al. (2013, see their Sect. 2) and includes wind-driven sea spray emissions (Guelle et al., 2001), prescribed emissions of mineral dust and volcanic sulfur (Dentener et al., 2006), terrestrial dimethyl sulfide (DMS; Spiro et al., 1992), and natural SOA precursors (Guenther et al., 1995), as well as prescribed anthropogenic and biomass burning emissions representative of the year 2000 (Lamarque et al., 2010). Emissions of the long-lived greenhouse gases (CO2 and CH4) are implicitly considered by relaxing their near-surface mixing ratios to observed values for the year 2000, based on data from the Advanced Global Atmospheric Gases Experiment (AGAGE) and the National Oceanic and Atmospheric Administration/Earth System Research Laboratory (NOAA/ESRL). This task is fulfilled by the submodel TNUDGE. For the details on the treatment of aerosol emissions in the model, we refer the reader to Righi et al. (2013). Here, we only describe the parts of the emissions setup that were updated or required MADE3-specific treatment.
Oceanic DMS emissions are calculated according to a formulation by Liss and Merlivat (1986) in ONEMIS, taking into account the dependence on wind speed and sea surface temperature. In comparison to Righi et al. (2013), we use a more recent climatological near-surface seawater DMS concentration dataset (Lana et al., 2011) as an input to this parameterization.
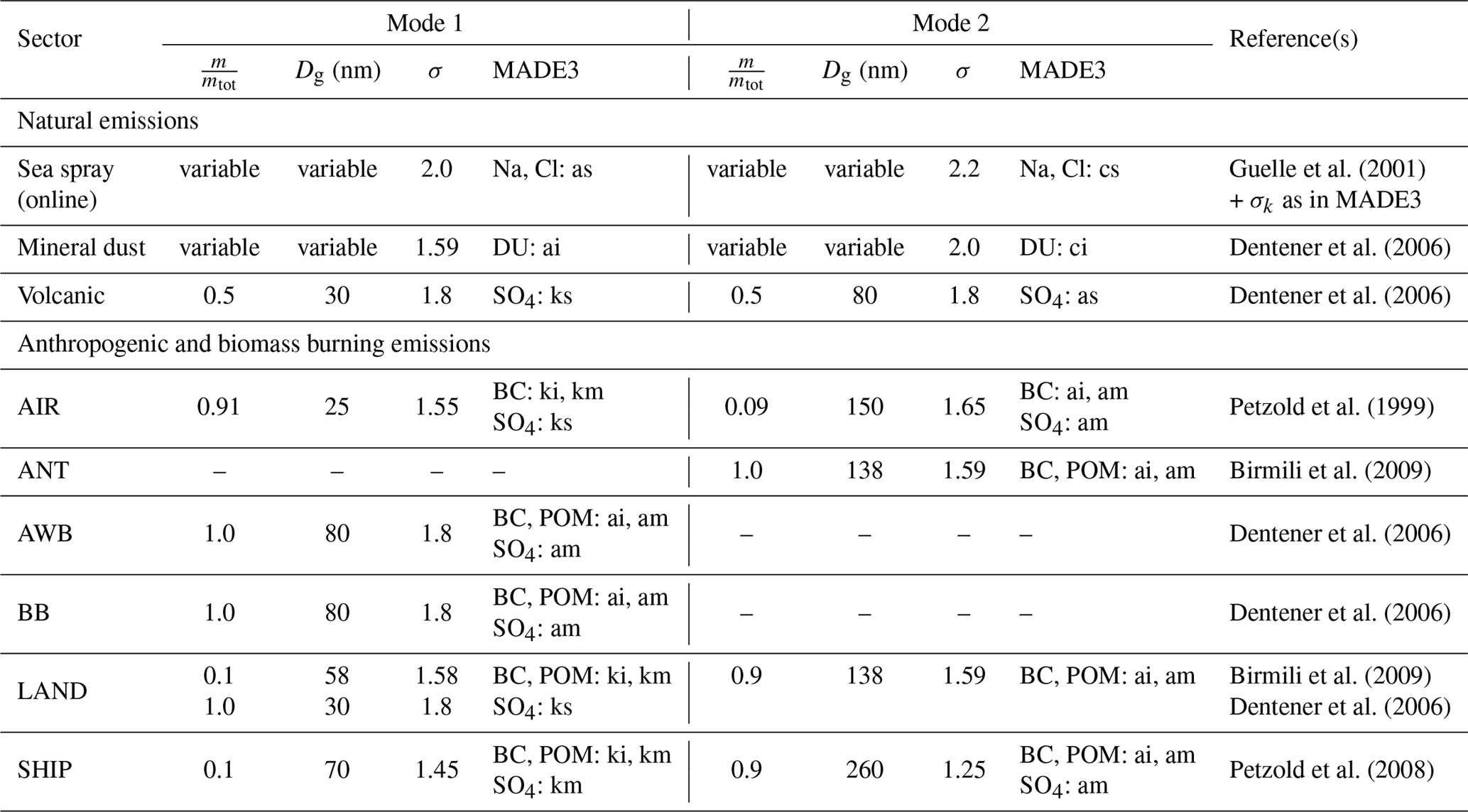
As aerosol particle number concentrations, size distributions, and mixing states are not included in most of the datasets that we use, we made typical assumptions for mixing states and size distributions here. Following Cooke et al. (1999) and Lohmann et al. (1999), we consider 80 % of the emitted BC and 50 % of the emitted POM to be hydrophobic and assign them to the insoluble MADE3 modes. Note that in the present setup only combustion sources of primary POM are considered and we assume that BC and POM are emitted as internal mixtures in the form of soot particles. Consequently, the hydrophilic fractions are assigned to the mixed modes. Depending on the emission sector, SO4 is either assigned to soluble or mixed modes. Where nucleation of ultrafine SO4 particles may play a role even in aged emission plumes, we assign these particles to the soluble Aitken mode. In the other cases, we assume that SO4 is efficiently scavenged by BC/POM particles and, consequently, choose a mixed mode. Volcanic SO4 is assigned exclusively to the soluble modes, as we do not consider any insoluble particles from volcanic emissions that could bear a coating. Mineral dust emissions are assigned to the insoluble modes, in which particles are assumed to be hydrophobic (Kaaden et al., 2009; Weinzierl et al., 2009), whereas sea spray emissions are assigned exclusively to the soluble modes. Unless explicitly specified in the datasets, i.e., for all emissions except those of sea spray and mineral dust, we derive number emissions from the mass emissions in analogy to the procedure employed by Righi et al. (2013). Under the updated assumptions for the size distribution parameters given in Table 2, the number emissions can thus be computed from the species mass emissions. These number emission fluxes are added to the corresponding MADE3 modes along with the mass emission fluxes from which they were derived.
Guelle et al. (2001)Dentener et al. (2006)Dentener et al. (2006)Petzold et al. (1999)Birmili et al. (2009)Dentener et al. (2006)Dentener et al. (2006)Birmili et al. (2009)Dentener et al. (2006)Petzold et al. (2008)Table 2Size distributions assumed for emitted particles. The term specifies the mass fraction provided by the respective measured or prescribed mode; Dg and σ specify, respectively, the median diameter and geometric standard deviation of the lognormal distributions used to describe the number size distributions of the modes. The “MADE3” columns show the species and modes to which the emissions are assigned. The mode naming convention follows Kaiser et al. (2014): one of the indices “k”, “a”, or “c” is used to specify the Aitken, accumulation, or coarse mode, respectively, and “s”, “m”, or “i” as a second index specifies the soluble, mixed, or insoluble mode, respectively. The value “variable” in the mass fraction and median diameter columns indicates that both number and mass emissions are provided by the emission data source, so that number emissions do not have to be derived from the mass emissions. The emission sectors are abbreviated as follows: “AIR” for aviation, “ANT” for anthropogenic non-traffic, “AWB” for agricultural waste burning, “BB” for biomass burning, “LAND” for land transport, and “SHIP” for shipping.
This section is organized as follows. First, we evaluate simulated near-surface aerosol particle mass concentrations by comparing them to measurements from four different station networks (Sect. 3.1). We then move on to a comparison of the vertical distribution of the simulated aerosol to aircraft measurements of BC mass mixing ratio and of particle number concentration (Sect. 3.2). In Sect. 3.3, we discuss comparisons of simulated size distributions to measurements. Subsequently, we present a method to compare global aerosol model output to size-resolved electron microscopy particle composition measurements, together with a first application (Sect. 3.4). As an aerosol measure derived from the vertical distribution of particle concentrations, composition, mixing state, and size distributions, we compare simulated AOD to satellite measurements and station network data (Sect. 3.5). Finally, we discuss global burdens of the simulated aerosol particle species along with the species' tropospheric residence times. Section 3.1–3.3 and 3.5 draw heavily on model evaluation with the help of the Earth System Model eValuation Tool (ESMValTool; Eyring et al., 2016).
When comparing global model output to observational data, several issues have to be taken into account. As Schutgens et al. (2016a) pointed out, differences of several tens of percent between simulations and measurements can arise simply due to spatial sampling, when comparing grid-box average values to localized observations. Furthermore, observed and simulated microphysical and chemical aerosol properties may not always be fully consistent. Many measurement methods, for instance, are only sensitive to a certain part of the atmospherically relevant particle size spectrum. Specifically, to date, fine-mode particles (diameters up to ∼1–2.5 µm) have received most attention in the literature. This is especially important when comparing “total” aerosol (species) mass and number concentrations. In addition, there is often not a one-to-one correspondence between simulated and measured aerosol species. A prominent issue in this context is related to measurements of “black carbon”, “soot”, “elemental carbon”, “equivalent black carbon”, and “refractory black carbon” which “synonymously refer to the most refractory and light-absorbing component of carbonaceous combustion particles” (Petzold et al., 2013). Different and partially inconsistent terminology has been, and is, used in the corresponding literature (Bond and Bergstrom, 2006; Petzold et al., 2013), which has to be kept in mind when comparing simulated BC concentrations to measurement data. Finally, some measurements may be inherently biased due to the method of particle sampling. According to Ames and Malm (2001), for instance, fine-mode NO3 may be underestimated in one station network (the Clean Air Status and Trends NETwork, CASTNET), while it could be overestimated in another (the Interagency Monitoring of PROtected Visual Environments network, IMPROVE). The results of the comparisons between MADE3 and the observations, as well as the possible reasons for specific biases, are discussed in the following.
The discussion here is mostly descriptive and only gives some possible reasons for deviations of simulations from observations. A thorough investigation of such discrepancies would require a large number of sensitivity simulations, including model experiments with different representations of processes and/or different spatial resolutions. Although beyond the scope of the present evaluation, this could be conducted as part of future studies, and also serve for quantification of simulation uncertainties.
3.1 Near-surface mass concentrations
Regular measurements within station networks provide both spatial and temporal data coverage that is well suited for evaluation of global aerosol models. Biases due to differences in timing of simulation output and observations (Schutgens et al., 2016b) are likely small in this context, as the measurements are typically taken by collecting particles on filters over several days and subsequently analyzing these samples.
The 10-year average simulated near-surface mass concentrations are compared here to the averages of available observational data in the period 1996–2005 from the following station networks: IMPROVE (Hand et al., 2011) and CASTNET (AMEC Environment & Infrastructure, 2015) in the US, the European Monitoring and Evaluation Programme network (EMEP; Hjellbrekke, 2014), and the Acid Deposition Monitoring Network in East Asia (EANET; Network Center for EANET, 2014). The simulation data are always the sum of the contributions from all modes in the lowermost model layer, i.e., up to ∼100 m. The comparison provides an indication of the model's ability to reproduce the climatological state of the Northern Hemisphere continental aerosol, where anthropogenic emissions are largest. As the emission dataset is assumed to represent year 2000 conditions (Sect. 2.4), the period for the observational data was chosen symmetric to the year 2000. Note, however, that most of the stations (across networks) do not provide complete temporal coverage of the years 1996–2005, which may lead to biases. Specifically, all EANET data that went into the comparison stem from the years after 2000. EMEP and EANET have fewer stations and can thus provide less data than IMPROVE and CASTNET (see Table 3).
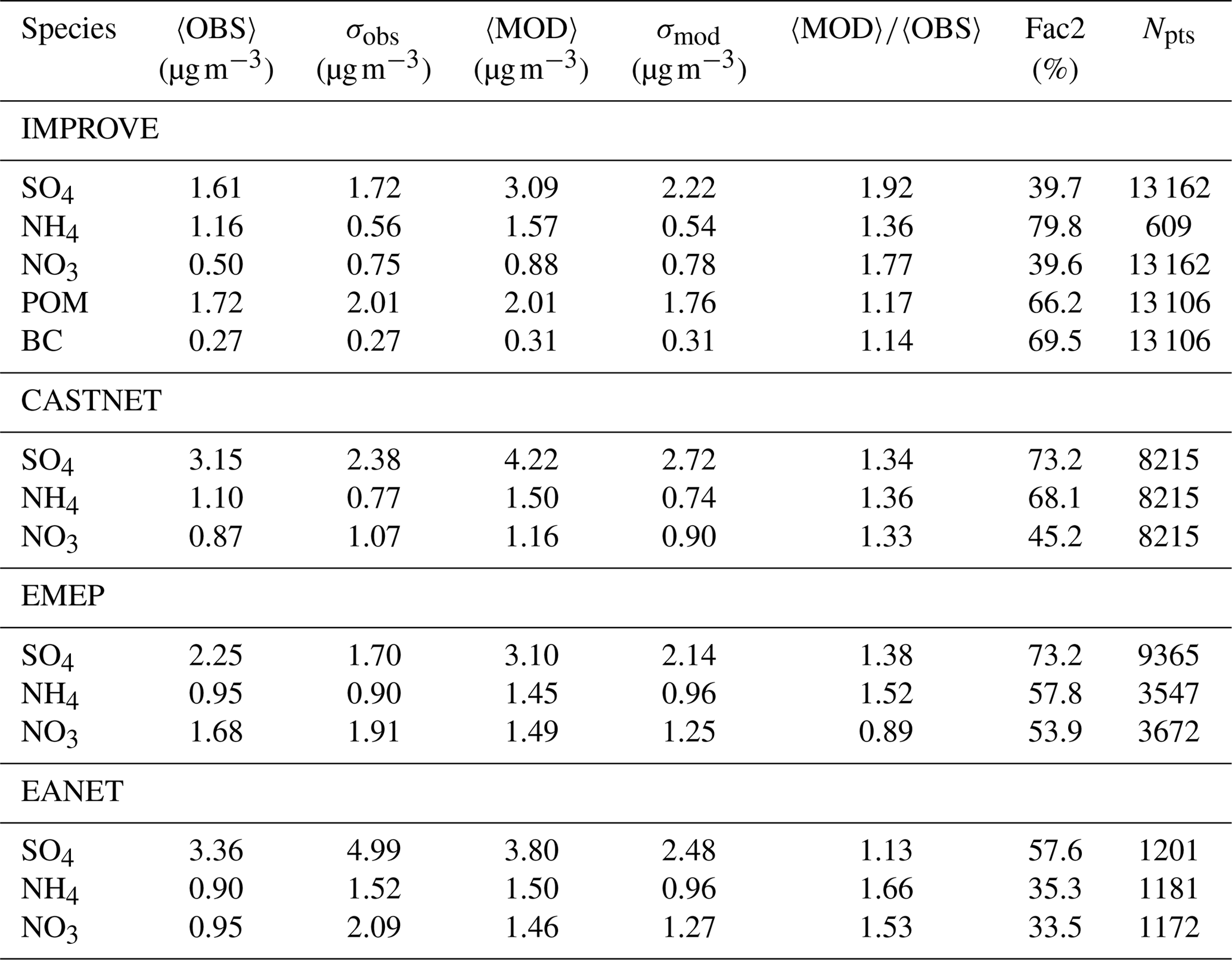
Table 3Summary of the model–observations statistical comparison of monthly mean near-surface aerosol concentrations from the EMAC simulation with MADE3 and station network data. 〈OBS〉 (〈MOD〉) stands for the arithmetic mean over all data points of the observations (simulation), σobs (σmod) for the standard deviations, Fac2 for the percentage of simulated values that are within a factor of 2 of the corresponding observational values (i.e., ), and Npts for the number of data points, i.e., monthly averages, that went into the comparison. See Appendix A2 in Righi et al. (2013) for more details about the methodology.
Concentrations of the secondary inorganic aerosol species SO4, NH4, and NO3 are the most widely measured and typically have the longest records, while BC and POM are only measured in the IMPROVE network. This subsection is ordered accordingly: the secondary species are discussed first and the (mostly) primary aerosol components thereafter.
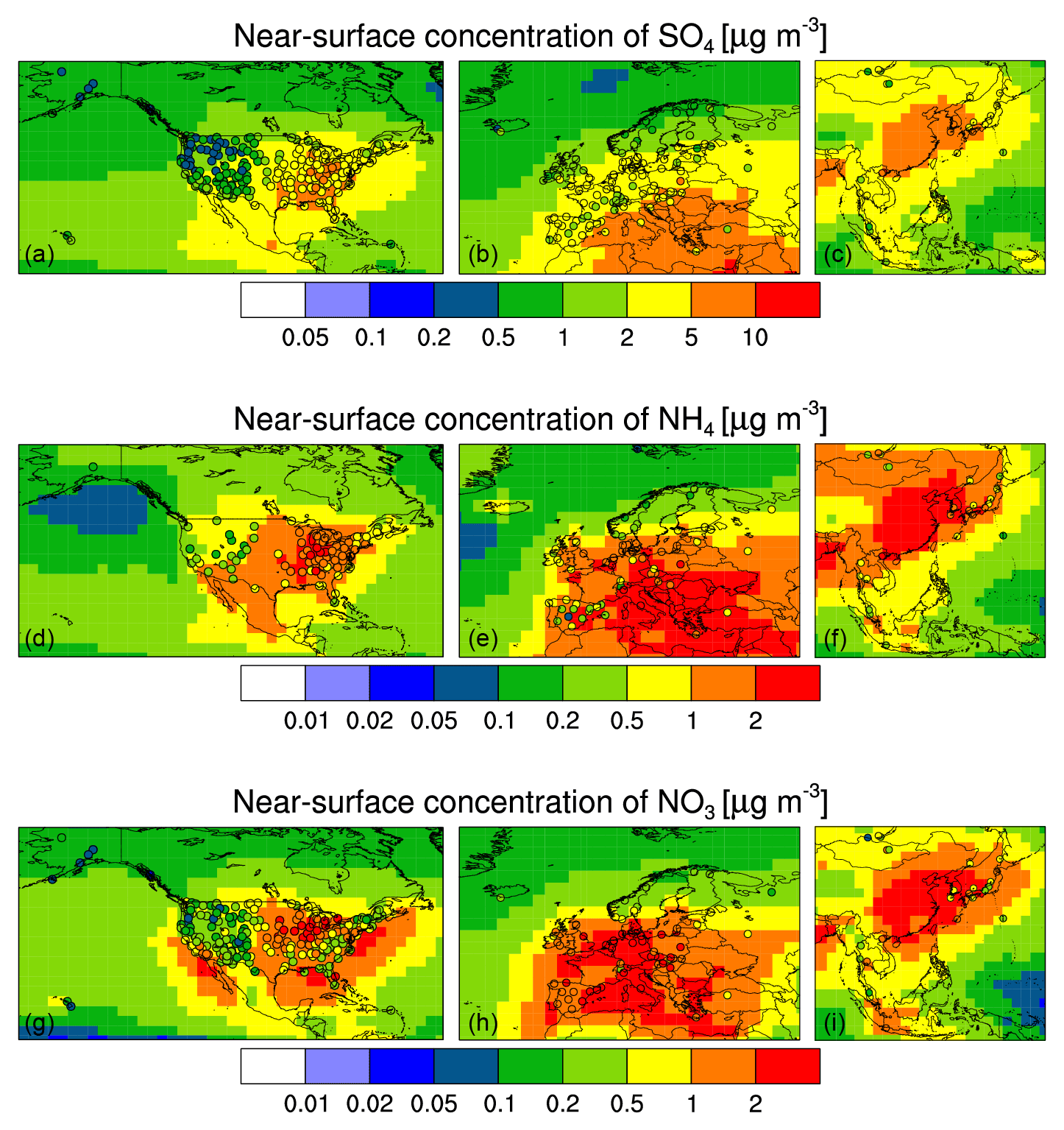
Figure 2EMAC simulation with MADE3 (background color; “pixels” correspond to the model grid) vs. observations from station networks (filled circles): multi-year average near-surface secondary inorganic aerosol mass concentrations, i.e., SO4 (a–c), NH4 (d–f), and NO3 (g–i). The observational data in the three panels of each row are drawn from CASTNET and IMPROVE (a, d, g), EMEP (b, e, h), and EANET (c, f, i).
The geographical distribution of near-surface SO4 concentrations (Fig. 2) is well reproduced over Europe and east Asia, albeit with a small bias (Table 3). Over the US, agreement between simulated and measured concentrations is better in the east than in the northwest. The model mostly reproduces the spatial pattern in this region, but it does not capture the west–east gradient seen in the observations, and is biased high. The relative deviation of near-surface SO4 concentrations is notably larger in the case of IMPROVE compared to the other networks (Table 3). As Ames and Malm (2001) do not find systematic differences in SO4 concentrations between co-located IMPROVE and CASTNET measurements, a possible reason for this difference lies in the locations of the IMPROVE stations. The relative deviations are largest in the northwestern part of the US (Fig. 2), where most of the stations used in the comparison are IMPROVE stations. These stations are mostly located in national parks and wilderness areas, i.e., in rather clean environments, whereas the large model grid boxes also cover more polluted areas in the vicinity of these areas.
Observed values of NH4 concentrations are spatially more heterogeneous than those of SO4 concentrations, down to scales that cannot be captured by the coarse resolution of the model. Furthermore, emissions of the NH4 precursor NH3 are much more uncertain and variable than those of SO4 precursors. That said, model performance is mostly similar for NH4 and SO4 (Fig. 2). This also means that the west–east gradient over the US is again underestimated. Note that, in the case of NH4, most IMPROVE stations with available data are located in the eastern part of the US, where agreement of the simulated concentrations with the observations is slightly better than in the west (see also Table 3). The north–south gradient over Europe is generally well captured, with the exception of Spain and the western Mediterranean.
The simulated near-surface NO3 concentrations agree remarkably well with the observations across Europe (Fig. 2). With respect to the IMPROVE data for the US, it must be noted that several tens of percent of the simulated NO3 mass belong to the coarse modes. In contrast to CASTNET, however, IMPROVE stations only sample particles up to a size of ∼2.6 µm (IMPROVE, 1995), so that deviations have to be expected, although partly compensated by the tendency of IMPROVE to overestimate fine-mode NO3. The comparison of NO3 concentrations to data from CASTNET and EANET yields similar results as for NH4.
Only IMPROVE stations routinely monitor carbonaceous aerosol components. Hence, the POM and BC simulation vs. observations comparison (Fig. 3) includes only data from the US. IMPROVE reports mass concentrations for organic carbon (OC), which were converted to POM by multiplying by a factor of 1.4 (Dentener et al., 2006). Both the magnitude and the spatial distribution of simulated near-surface concentrations generally agree well with the observations. This was expected, since the primary aerosol species POM and BC are only marginally affected by uncertainties associated with gas- and liquid-phase precursor chemistry and gas–particle partitioning which potentially cause distinct discrepancies in the case of secondary species.
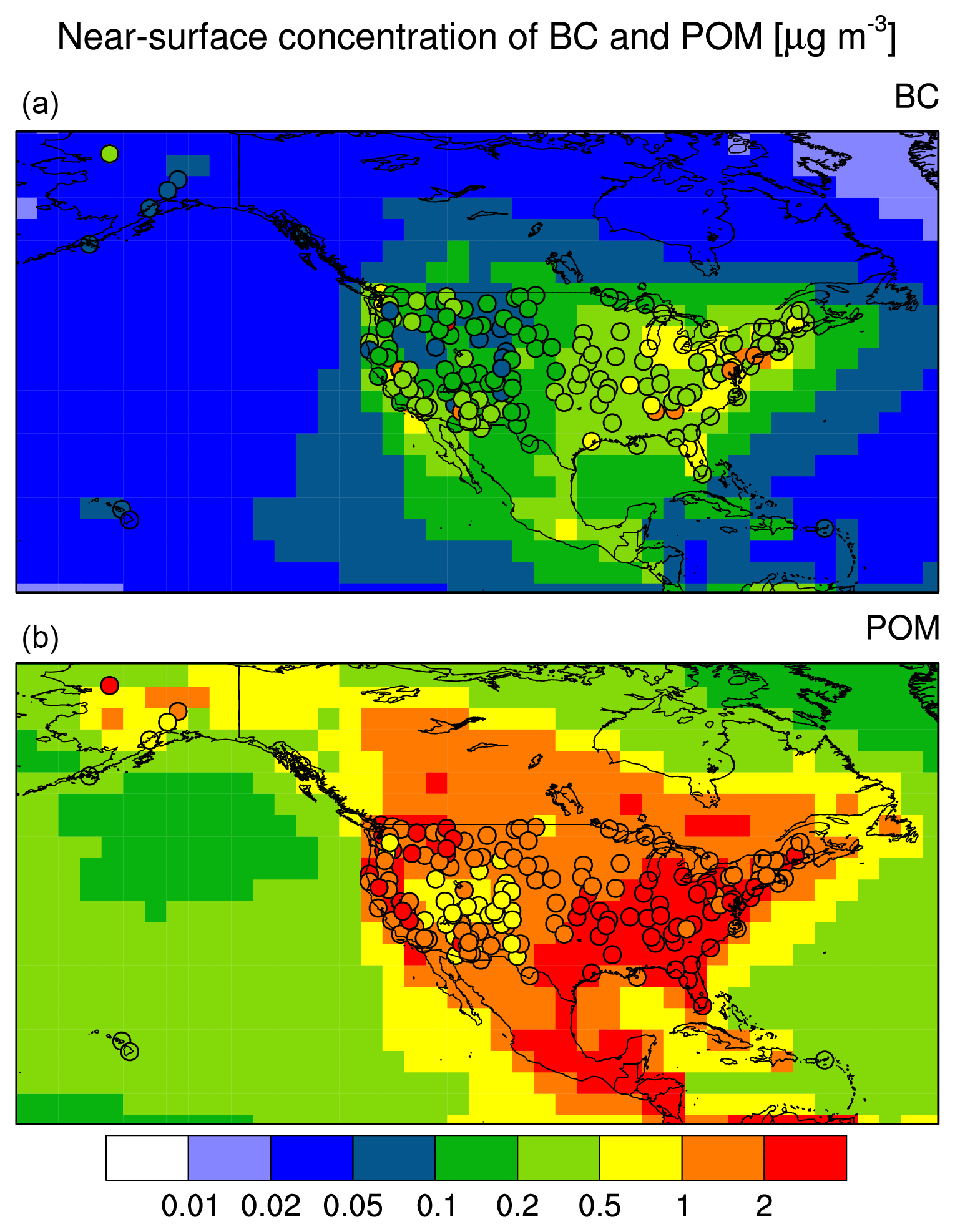
Figure 3EMAC simulation with MADE3 (background color; “pixels” correspond to the model grid) vs. observations from the station network IMPROVE (filled circles): near-surface BC (a) and POM (b) mass concentrations.
To evaluate dust concentrations, we follow the same strategy as Aquila et al. (2011), who compared simulated concentrations with the data based on a collection of measurements from 22 stations around the world in the context of the AEROsol Comparisons between Observations and Models (AEROCOM) project (Huneeus et al., 2011). The results are shown in Fig. 4: the data from the different stations are grouped in order of increasing dust load according to the respective average dust concentrations (1 to 7: low, 8 to 16: medium, and 17 to 22: high). EMAC (MADE3) generally underestimates dust concentrations, especially in comparison to the medium- and high-concentration stations, whereas the annual cycle is captured reasonably well. As pointed out by Aquila et al. (2011), this discrepancy could be due to the use of an offline monthly mean climatology for dust emissions in the model, rather than an online, wind-driven dust emission scheme. This could lead to a misrepresentation of atmospheric dust transport and removal. Furthermore, the climatology is representative of the year 2000, which was characterized by relatively low dust emissions. MADE3 simulations with more detailed dust emission parameterizations are planned as a subject of future studies. It is interesting to note, however, that the ability of MADE3 to reproduce dust concentrations has improved considerably over the previous MADE-in version of Aquila et al. (2011), who used the same input climatology for dust emissions.
The station networks also provide measurements of the sea spray components, i.e., Na and Cl. However, their concentrations are extremely low over the continents and, consequently, very sharp land–sea gradients in the near-surface concentrations of these species occur. These gradients cannot be accurately resolved by the model, which complicates the comparison with the observations, especially for stations in coastal areas. Hence, the station network data are not suited to evaluate sea spray aerosol. For an evaluation of the simulated marine aerosol, we refer to Sect. 3.5, where comparisons with satellite date are presented.
A comparison of simulated near-surface concentrations of various aerosol species to observations at high latitudes is challenging, since observational data for those regions are scarce and only a plausibility check could be performed here. Simulated BC concentrations are close to those measured over 2 years in North Greenland (as reported by Massling et al., 2015), while simulated SO4 concentrations are roughly a factor of 2 higher than the measured ones. Total aerosol mass concentrations over Antarctica are larger compared to measurements taken by Hara et al. (2014) during the austral summer 2007–2008.
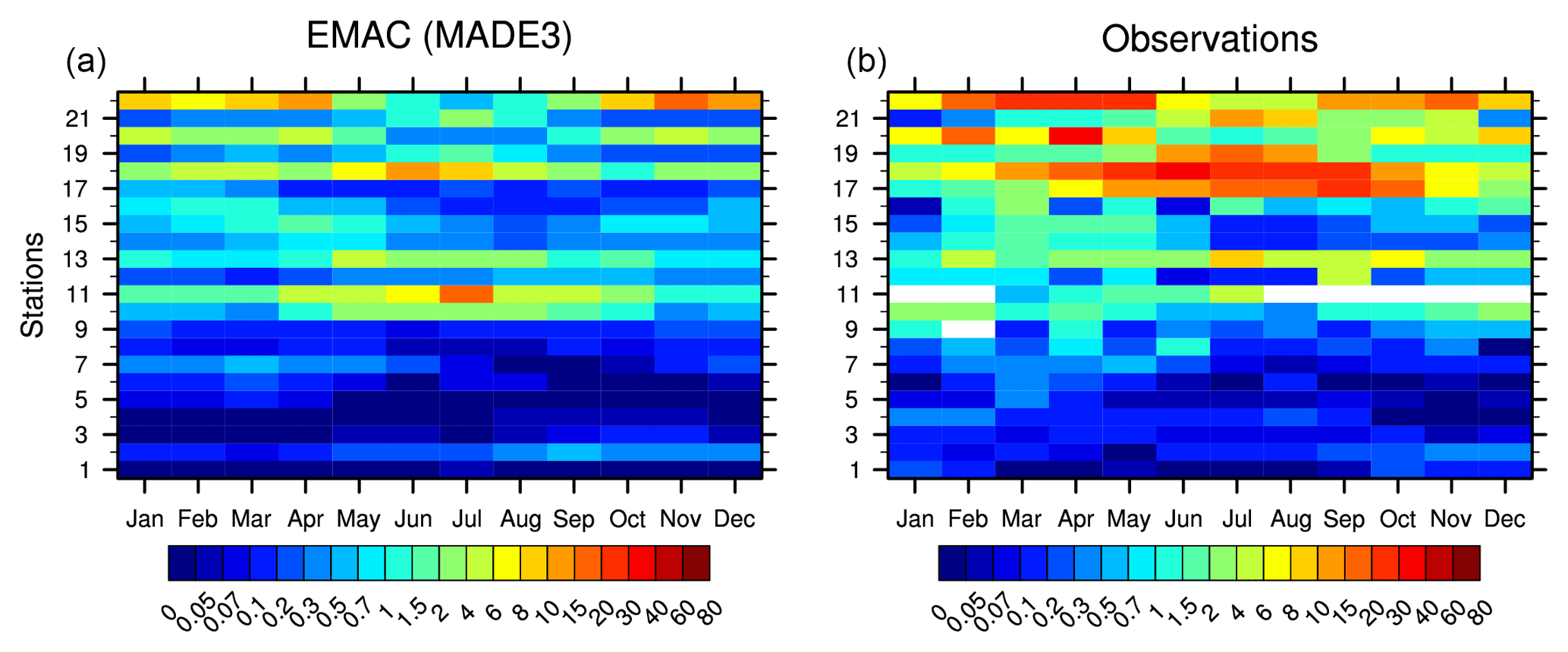
Figure 4Climatological annual cycle of simulated (a) and measured (b) dust surface-level concentrations in units of µg m−3. The observational data were collected by Huneeus et al. (2011). The locations of the 22 stations considered in the plot are shown in Fig. 6 of Aquila et al. (2011).
In conclusion, the simulated near-surface concentrations are mostly larger than the corresponding observed values (Table 3). This high bias is largest for the secondary aerosol components SO4, NH4, and NO3 but only small in the case of BC and POM. It may indicate a too-low efficiency of the deposition processes, which might also explain some of the discrepancies discussed in the next subsection that deals with vertical profiles of BC. That the overestimation is generally larger for soluble components indicates that cloud processing may also play a role in the deviations. That said, the statistics of our comparison with IMPROVE data are very similar to those obtained with a previous EMAC version using MADE-in (Aquila et al., 2011). The only exception here is NO3, which could not form on coarse-mode particles in MADE-in and therefore could not reach as high concentrations as in the present study. However, as mentioned above, IMPROVE stations do not provide any insight into NO3 concentrations associated with particles ≳2.6 µm.
The high bias of near-surface mass concentrations of secondary species found here is not typically seen in studies using other global aerosol models. Although (relative) discrepancies are often of similar magnitude to those obtained here, the deviations are typically more variable in their directions for different species (e.g., Bauer et al., 2008; Mann et al., 2010; Pozzer et al., 2012; Lee et al., 2015). For instance, EMAC (MADE3) simulates larger average sulfate concentrations than observed by all considered station networks. The corresponding biases amount to 13 %, 38 %, 34 %, and 92 % compared to EANET, EMEP, CASTNET, and IMPROVE, respectively. In contrast, Lee et al. (2015) found a similar high bias compared to IMPROVE (95 %) but a low bias compared to observations from European sites (−13 %). Other studies even show a general low bias. For example, the average sulfate concentrations obtained by Pozzer et al. (2012) show a low bias of −45 %, −16 %, and −28 % compared to EANET, EMEP, and CASTNET, respectively. Hence, EMAC (MADE3) shows a tendency towards enhanced sulfate concentrations. Nevertheless, the ability to simulate several tens of percent of monthly mean values within a factor of 2 of the observations indicates a quality of EMAC (MADE3) that is similar to that found in other model studies with this kind of analysis (Pozzer et al., 2012; Kirkevåg et al., 2013). It should also be mentioned that, in contrast to many other global aerosol models, EMAC (MADE3) performs quite well in the case of black and organic carbon. However, we note that the primary goal of the present study was not to improve on previous aerosol climatologies but rather to show that our new model, with its additional capabilities in terms of particle mixing state representation and coarse-mode particle interactions, also produces reasonable climatologies and hence is ready for investigating new topics that could not be addressed with the former versions of the model.
3.2 Vertical distribution
It is even more delicate to evaluate the global 3-D aerosol distribution than to evaluate the 2-D near-surface distribution. In contrast to the multi-year time series of measurements provided by surface station networks, aircraft measurements only sample aerosol along a specific flight trajectory. Hence, both the spatial and temporal data coverage is limited. Although arguably representative of the season and weather pattern during which flights took place, there is much more uncertainty associated with the comparison of climatological model output to aircraft measurements than with that to station network data. Nevertheless, aircraft campaigns provide a uniquely valuable way to measure vertical aerosol concentration profiles and are routinely used to evaluate the performance of global aerosol models in simulating the vertical aerosol distribution.
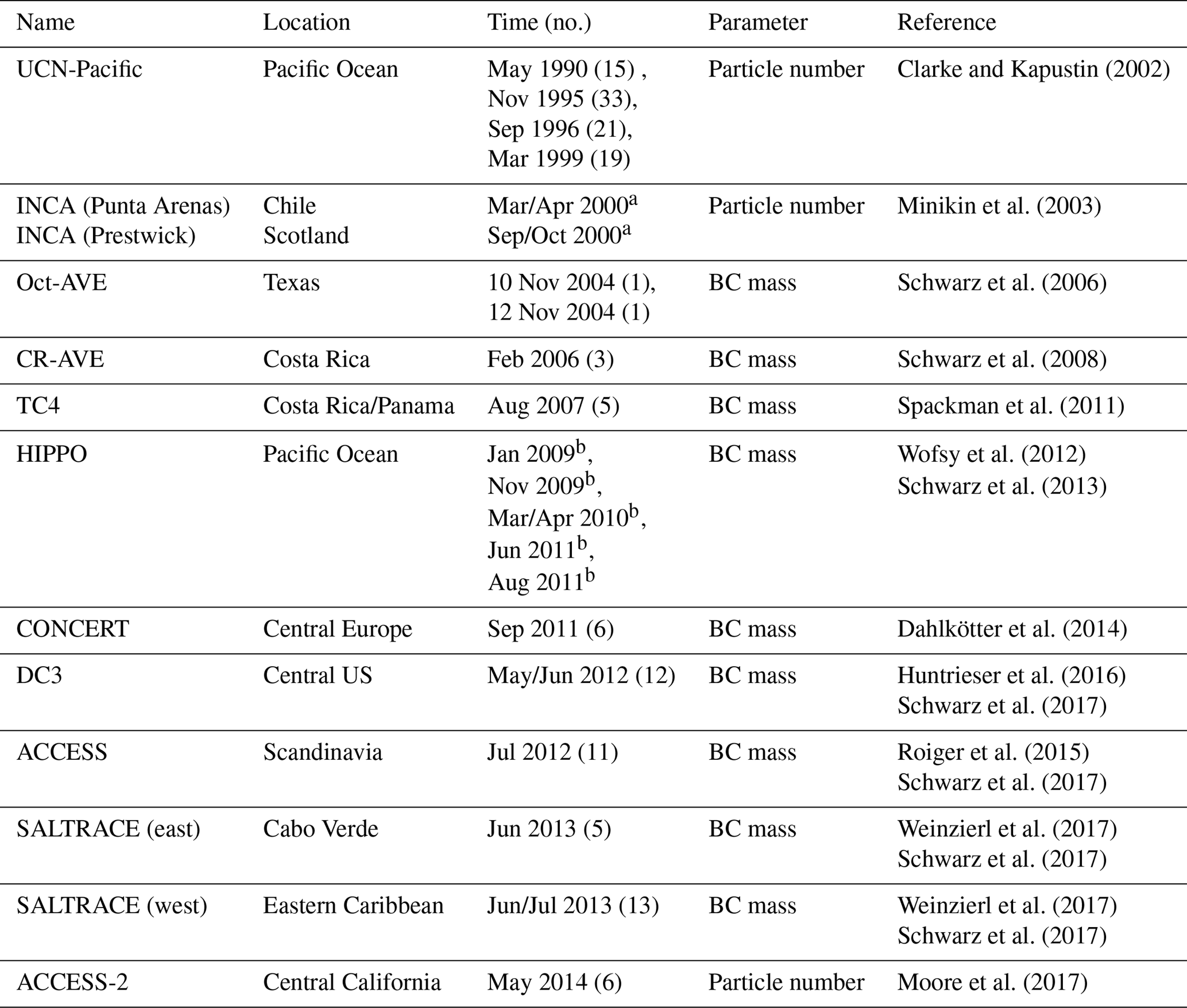
Here, we use observational data from campaigns between 1990 and 2014 over the Pacific Ocean, over North and South America, over Europe, and within the Saharan dust outflow over the Atlantic Ocean, as summarized in Table 4. Both BC mass mixing ratios (aerosol mass per unit mass of air; Fig. 5) as well as aerosol particle number concentrations (Fig. 6) were used. Depending on what a dataset provides, either mean values and standard deviations, or medians and percentiles, or both are compared between simulation and observations. Where data for individual flights are available, we show the median of each flight in the comparisons. The variability of the measured data includes spatial and temporal concentration variations during and between the flights. The variability of the model output, however, reflects the concentration variations around a climatological state, expressed by long-term mean or median concentrations, respectively. Simulation output data were selected from the grid boxes that include the flight trajectories and from the output time steps corresponding to the days or months of the year during which the flights took place (Table 4). This means that model data are selected for these days/months for each year of the simulation. Simulated meteorological-induced variability is captured well, even if only data for single days are extracted from each simulated year. For the comparisons, we vertically binned both the simulation and the measurement data into 1 km intervals. In situ GPS altitude was converted to ambient pressure using US standard atmosphere; this enabled the in situ to model comparison.
Clarke and Kapustin (2002)Minikin et al. (2003)Schwarz et al. (2006)Schwarz et al. (2008)Spackman et al. (2011)Wofsy et al. (2012)Schwarz et al. (2013)Dahlkötter et al. (2014)Huntrieser et al. (2016)Schwarz et al. (2017)Roiger et al. (2015)Schwarz et al. (2017)Weinzierl et al. (2017)Schwarz et al. (2017)Weinzierl et al. (2017)Schwarz et al. (2017)Moore et al. (2017)Table 4Summary of relevant details and references for the aircraft measurement datasets used in the evaluation of vertical aerosol profiles simulated with MADE3 in EMAC. The values in parentheses in the column “time” indicate the number of measurement flights considered for the evaluation.
a Total of 9–10 flights per campaign; numbers not given separately. b More than 700 profiles in total; numbers not given separately; number of flights not given.
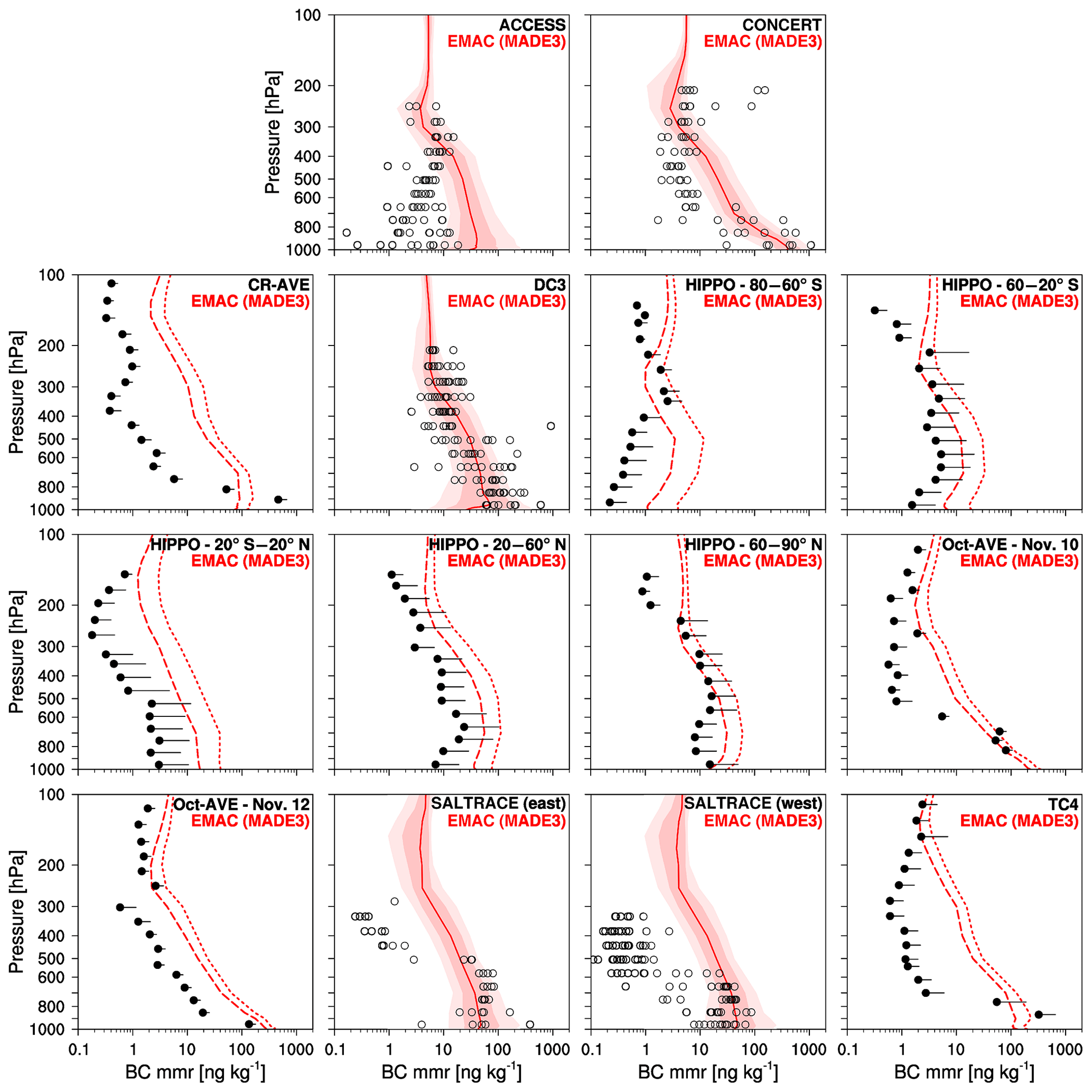
Figure 5BC mass mixing ratios (mmrs) in the EMAC simulation with MADE3 (red) vs. measurements during various field campaigns (black). Dashed lines and filled circles represent mean values; dotted lines and whiskers represent standard deviations, which are only shown in the direction of larger values for clarity. Solid lines stand for median values. Light and dark shadings indicate the 10th to 90th, and 25th to 75th percentiles, respectively. Hollow circles are the median values of individual flights. Descriptions of the campaigns are provided in Table 4 and in the text. Note that the vertical axis of the left plot in each row applies to the other plots of that row as well, and the horizontal axes of the plots in the lowermost row also apply to the plots in the other rows.
The general picture that emerges from the comparison of the BC mass mixing ratio profiles (Fig. 5), on the one hand, is a comparatively good resemblance of simulated and observed near-surface mass mixing ratios, particularly in polluted continental regions close to major BC sources. For instance, in the case of the DC3 and CONCERT campaigns, the simulated mixing ratios in the planetary boundary layer are close to the observed values. This is consistent with the similarity of the IMPROVE station measurements and the corresponding simulation results. On the other hand, the simulated near-surface mixing ratios in remote areas (e.g., the Pacific Ocean covered by the HIPPO campaigns), as well as those simulated for higher altitudes, mostly exceed the corresponding observations. A high bias of up to a factor of 10 occurs in many cases. In this context, it should be mentioned that the data obtained in missions initiated before 2003 used filter-based absorption measurements to infer effective BC concentrations, whereas missions initiated after 2003 used the Single Particle Soot Photometer (SP2) to report refractory BC concentration. The BC measurements with SP2 cover a particle size range of about 90–600 nm volume-equivalent diameter, assuming 1.8 g cm−3 void-free density, but for some datasets they have been slightly corrected (generally by <15 %) to reflect the total accumulation-mode BC mass. Except for fresh emissions very close to the sources, which most of the data do not represent, we expect the SP2 to capture most of the aerosol BC mass (Schwarz et al., 2006). Although underestimations of the total BC mass in any non-accumulation size modes due to the detection size limits in the measurements cannot be ruled out, discrepancies between model and observation of the order of a factor of 10, as found here, are probably almost certainly insensitive to this much smaller issue.
The high bias of the simulation with respect to the measured profiles could indicate an underestimation of aerosol scavenging as also hypothesized in the previous subsection. In addition, overestimated upward transport, possibly in convective plumes, could also contribute. Ouwersloot et al. (2015) found increased mixing ratios of an artificial tracer in the free troposphere when using an improved convective transport scheme that was recently developed for future versions of MESSy. This may mean that the general tendency of the simulated aerosol mass mixing ratios to exceed the observed values could actually be even larger, i.e., using a similar transport scheme here might lead to even larger discrepancies. Previous studies with MADE (MADE-in) rather showed a negative (slightly negative) bias of simulated vs. measured concentrations (Lauer, 2004; Aquila, 2009), which could indicate that the overestimation in the present work is caused outside the aerosol microphysics submodel, possibly by the representations of scavenging and vertical transport. However, the scavenging efficiency also depends on the aerosol size distribution (see Sect. 3.3), which is largely controlled by the aerosol microphysics submodel.

Figure 6Same as Fig. 5 but for aerosol particle number concentrations with various cutoff diameters.
Several other model studies included comparisons to the observational datasets used here. For instance, Lohmann et al. (2007, ECHAM5-HAM) achieved close agreement of the BC mass mixing ratio profiles for the Oct-AVE data but, using the same model with some modifications to aerosol-related mixed-phase cloud processes, Lohmann and Hoose (2009) found a similar overestimation of the CR-AVE data as that in Fig. 5. While Bauer et al. (2008, MATRIX) could better reproduce the decline of the BC mixing ratios with altitude close to the ground in the CR-AVE and TC4 data, EMAC with MADE3 performs better at higher altitudes. In a recent study, Lund et al. (2017) demonstrated that discrepancies between BC simulations with the OsloCTM2-M7 model and the HIPPO data can be strongly reduced by modifications in the model representation of BC wet scavenging. This again demonstrates that deficiencies in the model descriptions of wet removal can play a key role in this context. BC concentrations in the free troposphere are overestimated by many other models (e.g., Koch et al., 2009, 2010; Schwarz et al., 2013; Allen and Landuyt, 2014; Schwarz et al., 2017). Several authors, among them Kipling et al. (2013, HadGEM3-UKCA), Wang et al. (2013, CAM5), and Allen and Landuyt (2014, CAM5), found a better agreement with measured vertical profiles when improving the representation of aerosol–convection interactions. This includes aerosol activation, vertical transport, and wet removal in convective clouds. Note, however, that EMAC with MADE3 performs better in simulating upper tropospheric BC when compared to HIPPO data than the multi-model average of the models that took part in phase II of the AEROCOM model intercomparison project (Schwarz et al., 2013).
When comparing simulated aerosol particle number concentration profiles with measurements (Fig. 6), we find a comparatively good agreement over the Pacific Ocean, where both spatial and temporal coverage by the observations is most extensive (more than 200 profiles of the ultrafine condensation nuclei (UCN)-Pacific campaign; Clarke and Kapustin, 2002). Note that the simulation values are the result of an integration of the number size distribution from the cutoff diameter (3 nm) upwards. Especially in the Northern Hemisphere, the agreement is remarkable. In the equatorial latitudes, number concentrations agree well in the lower troposphere, but simulated number concentrations are smaller at high altitudes which could be a result of an underestimated efficiency of new particle formation. Over the southern Pacific, aerosol number concentrations are significantly underestimated in the lower troposphere, which could be an indication that especially natural sources of aerosol number are underrepresented in the model. For instance, new particle formation mechanisms including natural organic compounds (e.g., Kirkby et al., 2016; Tröstl et al., 2016) are neglected. Since natural precursors might be very relevant for new particle formation in the Southern Hemisphere where anthropogenic influences are comparatively small, this model deficiency might lead to particularly large discrepancies. The low bias of simulated aerosol number is not in contradiction to the high bias of the BC concentrations discussed above, since aerosol number is controlled by the large concentrations of ultrafine particles, which provide only very small contributions to aerosol mass and which are distinctively smaller than BC particles. Nevertheless, this could be an indication of a misrepresentation of the size distribution of such particles.
Similar to the comparison with the southern hemispheric UCN-Pacific data, the simulated lower tropospheric aerosol number concentrations are smaller than observed during INCA and ACCESS-2, which again could be a consequence of missing aerosol sources in the model. This deficiency can even affect the concentration of larger aerosol particles in the cloud condensation nuclei size range as reflected by the comparison with the INCA data. Hence, future work should focus on improving the representation of natural background aerosol, as also concluded by several other global aerosol modeling studies (e.g., Carslaw et al., 2013, 2017). In some cases, particularly in comparison to the INCA campaign, the model shows higher ultrafine particle number concentrations in the upper troposphere. A possible reason could be an overestimated nucleation rate. Zhang et al. (2012, ECHAM-HAM2) obtained a strong reduction in nucleation-mode number concentrations between ∼400 hPa and ∼150 hPa when switching from the Vehkamäki et al. (2002, 2013) scheme employed in MADE3 to a more recent parameterization.
Parts of the discrepancies discussed above could also result from temporal inconsistencies between the simulations and the observational data. We apply emission data for the year 2000, since a robust emission database is available for that year (Lamarque et al., 2010). These emissions are assumed valid for the years around 2000 (1996–2005). For consistency reasons, we adopt observational data from this time period in most of the comparisons discussed in this article. An exception is the data from recent aircraft-based field campaigns, which were carried out up to 14 years after 2000. However, deviations between model and observations in the more temporally dislocated cases are similar to those found for campaigns close to 2000. A systematic trend in the deviations does not occur. In addition, the deviations are clearly larger than the changes in emission rates occurring between 2000 and the years of the respective campaigns. Hence, internal model deficiencies, as described above, are probably the main reason for the deviations, rather than trends in the input data.
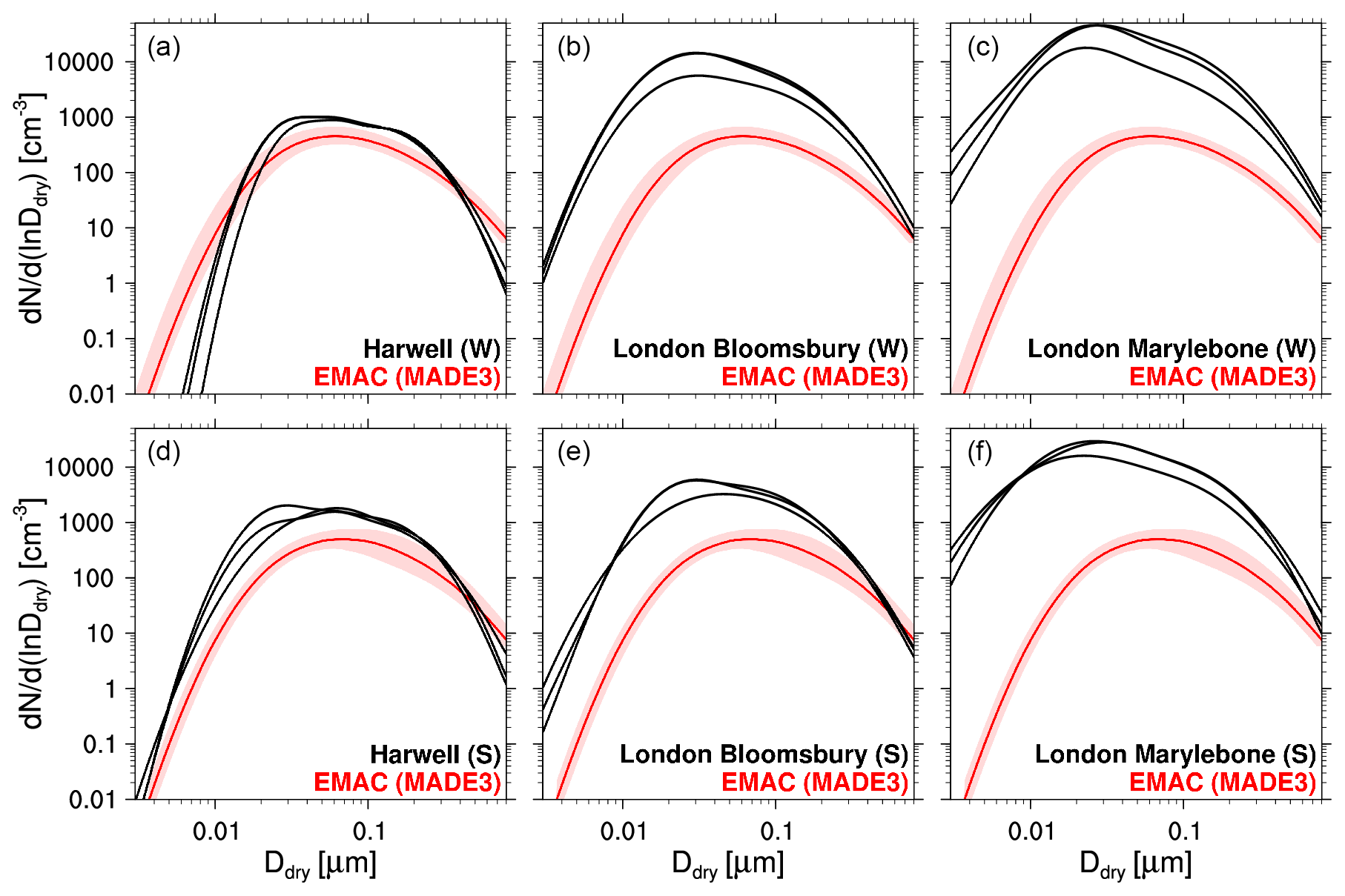
Figure 7Aerosol particle number size distributions in the EMAC simulation with MADE3 (red) vs. ground-based measurements (Putaud et al., 2003; Van Dingenen et al., 2004) during winter (W, a–c) and summer (S, d–f) at the same locations (columns). Each plot contains three measured size distributions: one for the morning, one for the afternoon, and one for the night hours. The three stations represent the following conditions (left to right): rural, urban, and kerb side (terminology adapted from Putaud et al., 2003). All measurement locations fall into the same model grid box, so that the simulated size distribution only differs between the top and bottom rows but not between columns. Solid lines stand for median values; shadings indicate the 25th to 75th percentiles. Note that the vertical axis of the left plot in each row applies to the other plots of that row as well, and the horizontal axes of the plots in the lowermost row also apply to the other plots in the respective columns.
3.3 Size distributions
Size distributions provide more detailed information on the aerosol population than integral particle number concentrations. Unfortunately, however, suitable observational data for our evaluation are scarce, especially when it comes to measurements above the ground. For the present study, we compared simulated size distributions to data from ground-based measurements (Putaud et al., 2003; Van Dingenen et al., 2004) and aircraft-based observations (Petzold et al., 2002). The latter have the particular advantage that size distributions were determined for different altitudes throughout the troposphere. Simulated size distributions are taken from the grid boxes corresponding to the geographical coordinates of the measurements and only from those time steps (in each simulated year) that correspond to the days or months of the observations. We found that variability due to model meteorology is captured well with this approach.
The ground-based measurements were performed at 10 European stations that monitored aerosol particle size distribution during at least one full season, i.e., either winter (December, January, February) or summer (June, July, August) in the 1990s or early 2000s. Putaud et al. (2003) fitted up to three lognormal modes to the measured distributions for three times of the day, namely the morning, the afternoon, and the night. Figure 7 shows a subset of our comparisons, which serves to illustrate our results and the problems associated with this type of evaluation.
Our main conclusion here is that the comparability of simulated and measured size distributions can be strongly affected by the specific characteristics of the local environments at the respective stations. This is especially evident when comparing simulation output to data from three stations that fall into the same model grid box, as we do in Fig. 7. Only one size distribution can be realized in this grid box at any given time in the model. While the model agrees comparatively well with the measurements at the rural station (Fig. 7a, d), it shows distinctively smaller concentrations for all particle sizes when compared to measurements from the urban background station (Fig. 7b, e) and the kerb-side station (Fig. 7c, f). This was expected, since local concentration enhancements occurring close to local sources cannot be resolved by the model with its large horizontal resolution of about 300 km. The rural station (Harwell) might occasionally be influenced by urban pollution since it is located in the vicinity of London. Hence, the long-term median concentration at this station is expected to be closer to the large-scale median concentration of the model grid box than the median values from the other two stations, as the grid box contains both urban and rural environments. This interpretation is consistent with comparisons to measurements at natural background stations (other grid boxes; not shown) where the model shows larger average concentrations than the observations which are expected to be representative of the less polluted fractions of the respective grid boxes.
Simulated near-surface size distributions over Europe appear to be strongly affected by the emissions. Hence, as most (prescribed) emissions in our simulation are considered as monthly averages, we see little variability of the size distributions. Another deficit of the MADE3 aerosol particle size distribution is the MADE3 output appearing almost unimodal in many cases, whereas the observations often show two or more distinct modes. This finding is consistent with the result of the box model test of MADE3 (Kaiser et al., 2014), and we now find it to be independent of season and location. The discrepancy may be caused partially by differences between the size distributions assumed to calculate particle number emission rates and the size distributions obtained after assigning these emissions to the respective MADE3 modes (Sect. 2.4). In most cases, the widths of the modes that were fit to the measured data are narrower than those assumed in MADE3, where σ=1.7 and σ=2.0 for the Aitken and accumulation modes, respectively. Simulations with alternative assumptions on mode widths are intended to be the subject of future studies.
Further possible contributions to deviations between the simulation and the observations could be related to the timing of simulation output and measurements as well as to the new particle formation approach employed in MADE3. The lack of temporal collocation of simulation output with measurement times may already bias our results (Schutgens et al., 2016b). In addition, Lee et al. (2013a, b) found that boundary layer nucleation of new particles could contribute up to several tens of percent to the uncertainty in number concentrations of particles larger than 50 nm. MADE3 includes an empirical nucleation scheme (Vehkamäki et al., 2002, 2013). As several model studies (e.g., Spracklen et al., 2006; Matsui et al., 2013; Makkonen et al., 2014; Pietikäinen et al., 2014) suggest, the incorporation of more advanced nucleation schemes can lead to a more accurate reproduction of observed aerosol particle number concentrations.
Despite the mentioned shortcomings, EMAC performs reasonably well with MADE3 compared to the published results of other global model–observation comparisons of aerosol particle size distributions. Neither Wang et al. (2009, CAM3-IMPACT) nor Zhang et al. (2012, ECHAM5-HAM2) achieved substantially closer agreement with their models. Even Lee et al. (2015, ModelE2-TOMAS) found similar deviations despite using a sectional model, which would be expected to resolve size distributions better than modal models with fixed mode widths.
Many of the arguments presented above also apply to the comparison of our simulation data to data from the LACE campaign (Fig. 8). The measurements were taken during July and August 1998 at different altitudes over northeastern Germany (Petzold et al., 2002). We use three-mode fits to the measured size distributions for four to five individual flights here, depending on the flight altitude. Again, the fitted modes are much narrower (σ≤1.6) than the MADE3 modes.
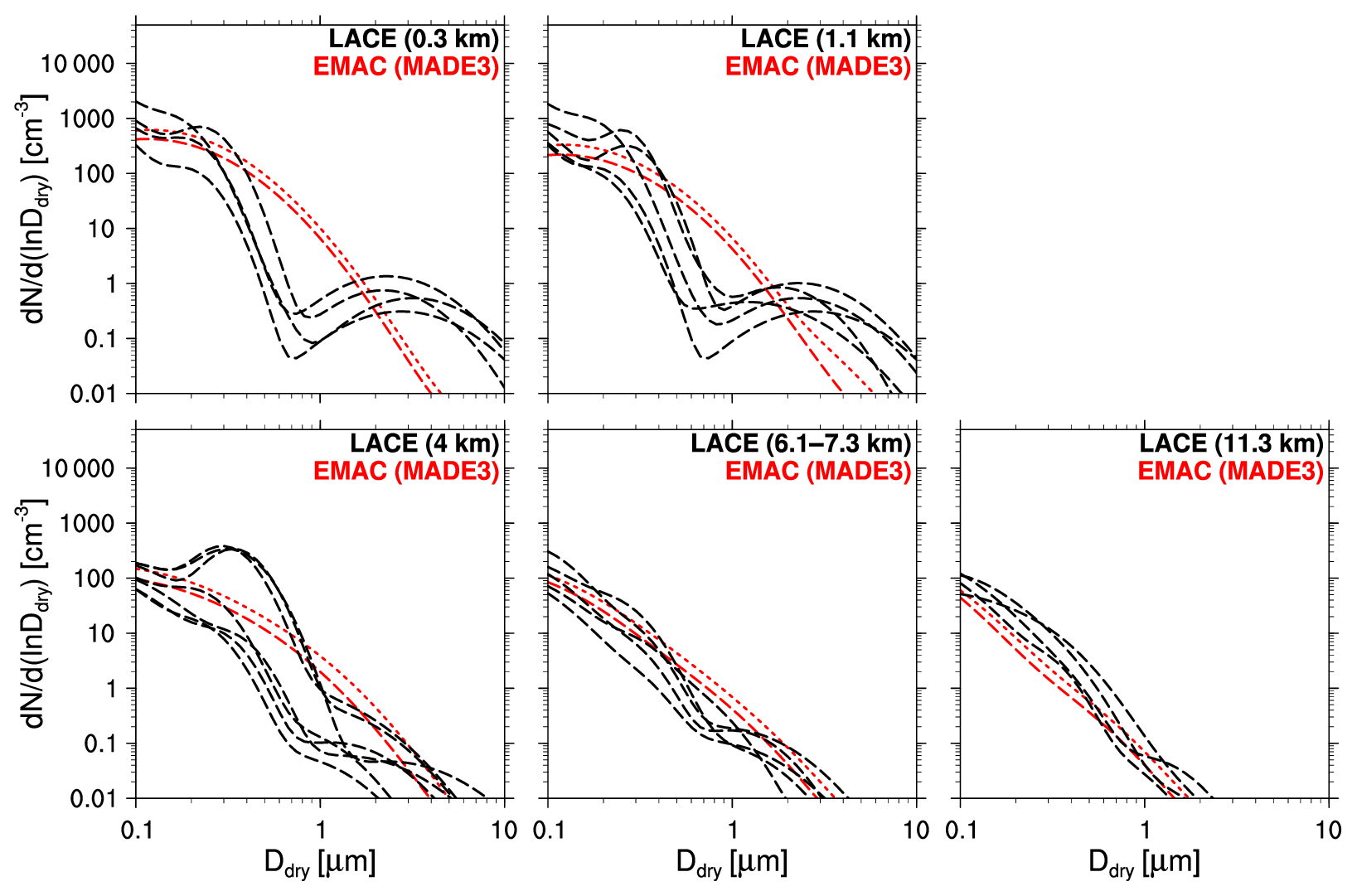
Figure 8Aerosol particle size distributions in the reference simulation with MADE3 (red) vs. measurements (black) for four to five individual flights (depending on altitude) during the LACE campaign over northeastern Germany. Dashed lines represent mean values; dotted lines represent standard deviations, which are only shown in the direction of larger values for clarity.
Notable differences between simulation and LACE data include the lack of the coarse mode at lower altitudes and the accumulation mode peak in the upper boundary layer/lower free troposphere from the simulation output. As the measured coarse mode declines with altitude, it may have to do with local, anthropogenically induced dust emissions that are not included in the emission dataset used here. The peak at ∼ 300 nm in the upper boundary layer/lower free troposphere measurements was caused by a forest fire aerosol layer that cannot be reproduced in the simulation because this specific fire is not contained in the emission dataset.
Looking at the remaining parts of the size spectrum and considering the model's capabilities, we see good agreement of the simulated size distributions with the LACE data. Furthermore, we find that agreement improves with altitude, i.e., with increasing particle age.
3.4 Size-resolved composition
To enable a specific evaluation of the new coarse-mode particle representation in MADE3, it is useful to compare model output to size-resolved particle composition measurements. However, such data rarely include coarse-mode particles, and the correspondence between simulated and measured quantities is not always straightforward. We therefore present a strategy for evaluating simulated size-resolved aerosol composition with the help of electron microscopy data of in situ sampled aerosol particles. For an initial application of this strategy, we chose a dataset from measurements performed in January and February 2008 at a ground station at Praia, Cabo Verde, by Kandler et al. (2011) during the SAharan Mineral dUst experiMent 2 (SAMUM-2) field campaign (Ansmann et al., 2011).
Particle sizes as determined in the electron microscopy measurements are given as equivalent circle diameters of the particles' projected areas. We assume that these can be directly compared to the diameters derived from the simulated aerosol particle number and component mass concentrations, the mode widths, and the assumed component densities, since spherical particles are assumed in the model (Kaiser et al., 2014).
The experimental analysis is performed on individual particles, i.e., 48 599 particles in the dataset used here. Overall, 13 major elements were detected in the investigated particle population. Based on the relative contributions of the elements to the particle volume, each particle is assigned to one of 12 different particle classes, e.g., sulfates, chlorides, oxides, and silicates (see Kandler et al., 2011, for details).
For the comparison to model output, this procedure has a severe drawback. It would require classification of the MADE3 particles according to the same, or analogous, rules as the measured particles. However, since all particles within each MADE3 aerosol mode are assumed to have the same composition (model assumption of perfect internal mixture of all involved compounds), only particles of a maximum of nine different compositions can coexist at the same time in each grid box of the model. Classification of model particles, or rather modes, is therefore not reliable from a statistical point of view. For instance, consider a mode that contains both SO4 and Cl. With the model assumption of perfect internal mixtures, its total volume can always be assigned to only one class, either to the sulfates or to the chlorides. In reality, however, the mode would likely contain both particles with a major contribution from sulfate (assigned to the sulfate class by the measurements) and particles with a major contribution from chloride (assigned to the chloride class by the measurements). Hence, classification of the model modes would create unacceptable sampling biases.
Furthermore, nitrogen compounds only produce weak signals in the measurements, and material from the sampling substrates can affect the analysis of carbonaceous matter. Of the species simulated by MADE3, only SO4, Na, Cl, and DU can therefore be determined reliably in the measurements of contributions to particle composition.
For these reasons, we adopted a different view on the electron microscopy data. In the approach employed here, the component masses of each analyzed particle are assigned to one of five diameter “bins” according to the particle's size. Only those components that can be compared to model output are considered. The chloride fraction is measured directly and considered to be derived exclusively from sea spray. The sodium fraction is also measured directly, but for correspondence to the MADE3 Na tracer (which represents the whole non-chloride sea spray mass in the model), the sea spray sulfate fraction has to be added. The latter can be derived from the measured chloride fraction under the assumption of a typical sea spray composition, i.e., 54.6 % of chlorine atoms, and 2.82 % of sulfur atoms, and under the assumption that all this sulfur is present in the form of sulfate. The rest of the detected sulfur is also assumed to stem from sulfate and can be compared to the MADE3 SO4 tracer. The mineral dust contribution is derived from multiple elements that are typical of mineral dust (silicon, aluminum, iron, magnesium, calcium, potassium, phosphorus, titanium, and sodium).
Model output is binned into the same diameter intervals as the measurement data by integrating the mass size distribution of each mode from the lower to the upper bin boundary and then summing up the contributions of the individual modes. Thus, measurement data and model output are brought to the same format and can be compared.
An example comparison is shown in Fig. 9. The measurement panel (Fig. 9b) displays the average particle composition over the whole SAMUM-2 campaign (26 individual days or 48 205 particles). The rest of the analyzed particles fell outside the size range presented here. With 3729 particles, the rightmost bin has the smallest database. For the model plot, 12-hourly output from the grid box that contains the measurement station was averaged over the 10 evaluated simulation years, considering only the days of the year when the measurements took place.
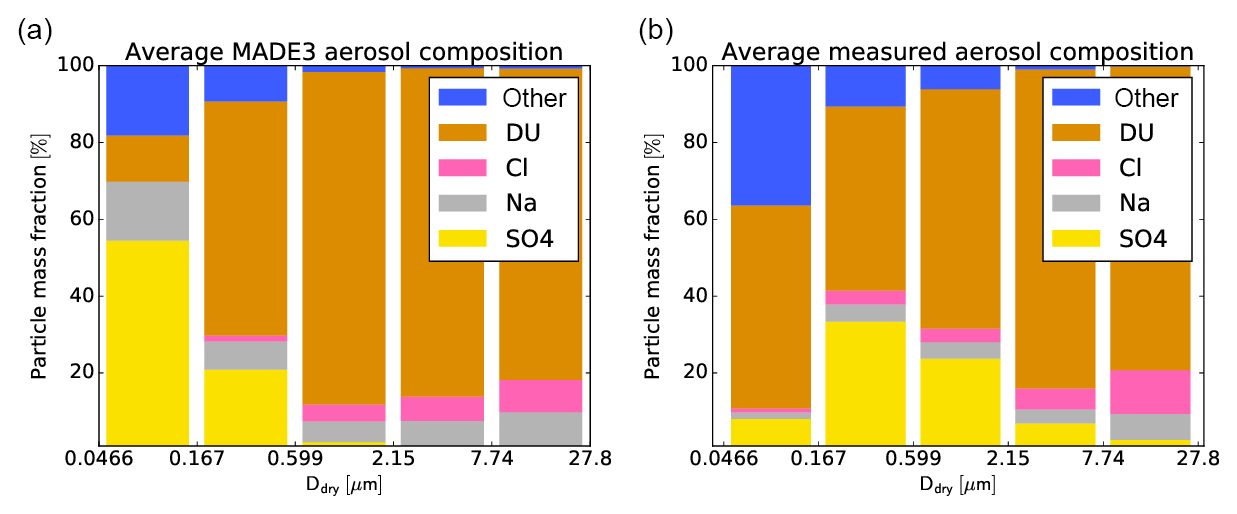
Figure 9Average size-resolved aerosol composition as simulated by the model (a) and as measured during the SAMUM-2 campaign (b). Only the mass fractions of species that can be compared between measurement data and model output are depicted.
The result shown in Fig. 9 must be interpreted with caution. It is not possible to exactly reproduce the conditions during the SAMUM-2 campaign with the model setup used here, except by chance. Especially, the monthly mean year 2000 DU and SO4 emissions in the simulation may not be representative of the actual situation in the beginning of the year 2008. Moreover, the meteorological features of the simulated years (1996–2005), which largely impact the simulated mean aerosol properties at the measurement site, might not correspond well to the specific meteorological conditions in 2008. Local pollution sources cannot be resolved by the model either. That said, the comparison reveals similarities between the simulated and measured data in the decrease of the SO4 fraction and the increases of the sea spray (Na plus Cl) and DU fractions with increasing size. Major discrepancies, however, exist in the composition of the smallest compared particles. We also analyzed the model biases in the individual years, but the interannual variability (not shown) was found to be small; hence, meteorology alone cannot explain the discrepancies. Model misrepresentations, for instance, of the mineral dust particle size distribution, the local sulfate concentration, or the competition between nucleation and condensation of gaseous H2SO4 could also play a role. On the other hand, the electron microscopy data analysis in particular of the smallest size fraction might have a bias towards an underestimation of sulfate particles due to their instability under the electron beam. Since the number concentration of particles in this size fraction is comparatively high, a thorough analysis, including comparisons of the measured and simulated size distributions and also measurement uncertainties, should be the subject of a separate study.
3.5 Aerosol optical depth
AOD provides an integral measure of the vertical aerosol column. On the one hand, it can be computed from the simulated aerosol properties discussed in the previous subsections, i.e., particle composition, particle sizes, and their vertical distributions. On the other hand, AOD can also be derived from measurements with ground-based and satellite-borne radiometers. Here, in Fig. 10, we compare the simulated AOD to data from the ground-based AErosol RObotic NETwork (AERONET; Holben et al., 1998, 2001) and against satellite data from the European Space Agency Climate Change Initiative (ESACCI) Swansea University (SU) Along-Track Scanning Radiometer 2 (ATSR-2) v4.21 aerosol product (North et al., 1999; Bevan et al., 2012; Holzer-Popp et al., 2013; de Leeuw et al., 2015) and from the MODerate resolution Imaging Spectroradiometer (MODIS) Level 3 Collection 6 data (Levy et al., 2013). Since annual mean AOD regionally changed by up to 10 % during the last decade (2000–2010, e.g., Yoon et al., 2014; Pozzer et al., 2015), only the year 2000 data are used in the comparisons of annual mean AOD here. This does not apply to MODIS, for which we considered the year 2003, i.e., the earliest year available in the time period covered by the instrument.

Figure 10Annual mean AOD in the reference simulation with MADE3 (a, background color) vs. observations from the AERONET network (a, filled circles) and vs. satellite data from the ESACCI Swansea University (SU) ATSR-2 v4.21 aerosol product (b, c) and from MODIS Level 3 Collection 6 (d, e). The comparison against satellite data is shown as both absolute (b, d) and relative (c, e) difference. “Pixels” in the panels correspond to the model grid. Results are shown for the year 2000 (AERONET and ESACCI) and for 2003 (MODIS).
The results shown in Fig. 10 reveal that, in comparison to the measurements, the model simulates up to ∼50 % higher AOD in the major pollution and biomass burning plumes that originate in east Asia, central Africa, and South America. This high bias is consistent with the general tendency of the model to overestimate aerosol mass concentrations as seen in Sect. 3.1 and 3.2. These high mass concentrations may also entail a higher aerosol water content, which could increase AOD further. The sensitivity of AOD to differences in hygroscopic growth, i.e., water uptake due to aerosol particle hygroscopicity, was recently demonstrated, e.g., by Li et al. (2014). Furthermore, the simulated AOD can be very sensitive to the scavenging scheme for aerosol particles entrained into convective clouds. The choice of the scheme can lead to several tens of percent different annual mean AOD values (Croft et al., 2012). A better agreement is found in the case of less polluted areas such as the remote oceans. The sign of deviations occurring in these areas shows spatial variations. This could be an indication for misrepresentations of either natural aerosol sources or long-range transport of anthropogenic particles and should be the subject of further investigations in future studies. The model tends to underestimate AOD where DU is abundant, especially over the Sahara and the Arabian Peninsula. Potential reasons for this underestimation include the use of prescribed monthly mean year 2000 DU emissions, the assumption on the DU size distribution upon emission, and the DU representation in AEROPT, the submodel that computes aerosol optical properties (Sect. 2.1). Johnson et al. (2012) and Nabat et al. (2012) found improved agreement of simulated AOD with observations when using a parameterization with more of the emitted DU mass in the coarse mode. Furthermore, the studies by Zhao et al. (2013) and Mahowald et al. (2014) indicated that a modal representation of DU particles with fixed mode widths may have unavoidable shortcomings.
AOD data from different satellite instruments do not agree perfectly but show similar patterns in large parts of the globe. Consistent patterns of the two satellite–model differences may therefore indicate areas where the model could be biased. Remaining satellite–satellite differences do even occur for data from the same instruments, if they are obtained with different retrieval algorithms (e.g., Popp et al., 2016). Hence, one cannot expect perfect agreement of simulated AOD with the observations either. It has, for example, been shown that MODIS AOD is larger by about 0.03 on global average than ESACCI SU values, while the ESACCI SU algorithm significantly overestimates AOD in dust regions such as the Sahara (Lauer et al., 2017). Furthermore, uncertainties involved in the model calculations of particle optical properties can of course also contribute to deviations. While different models have different strengths and weaknesses, it is interesting to note that many models have a low bias in AOD on a global annual average basis (e.g., Pozzer et al., 2012; Kirkevåg et al., 2013; van Noije et al., 2014; Lee et al., 2015; Michou et al., 2015) rather than a tendency towards a high bias as seen here: 18 % (−5 %) with respect to the ESACCI (MODIS) data. Relative underestimations in some of the mentioned studies are actually larger than these values, so that we can claim reasonable performance of EMAC with MADE3 as a global aerosol model. Deviations of the order of 10 % should not be overinterpreted anyway, as the lack of temporal collocation of the simulation output with the measurement times can already lead to biases of this magnitude (Schutgens et al., 2016b).
3.6 Tropospheric burdens and residence times
Although it is not an evaluation in the sense of a check against observational data, a comparison of global tropospheric aerosol burdens and residence times to estimates from other model studies is also instructive. The burden mtot,a of aerosol species a is computed here as the sum over the volume integrals of the mass concentrations ca in all grid boxes. The simulated burdens are presented in Fig. 11 along with results from other modeling studies. The species' residence times, tres,a, can be derived from the burdens and the sums of the deposition fluxes, Fdep,a, as
In the case of primary aerosol species, tres,a can alternatively be estimated from the global emission fluxes Femis,a. Since emissions are the only source of primary species, it can be assumed that in an equilibrated global aerosol budget. Due to the short lifetime of tropospheric aerosol, this equilibrium assumption is well applicable here. Since Fdep,a can not yet be quantified in our current model version, we use Femis,a instead of Fdep,a in Eq. (1) to estimate tres,a for the primary species DU, BC, and POM. The quantification of tres,a for other aerosol constituents is intended to be the subject of future studies.
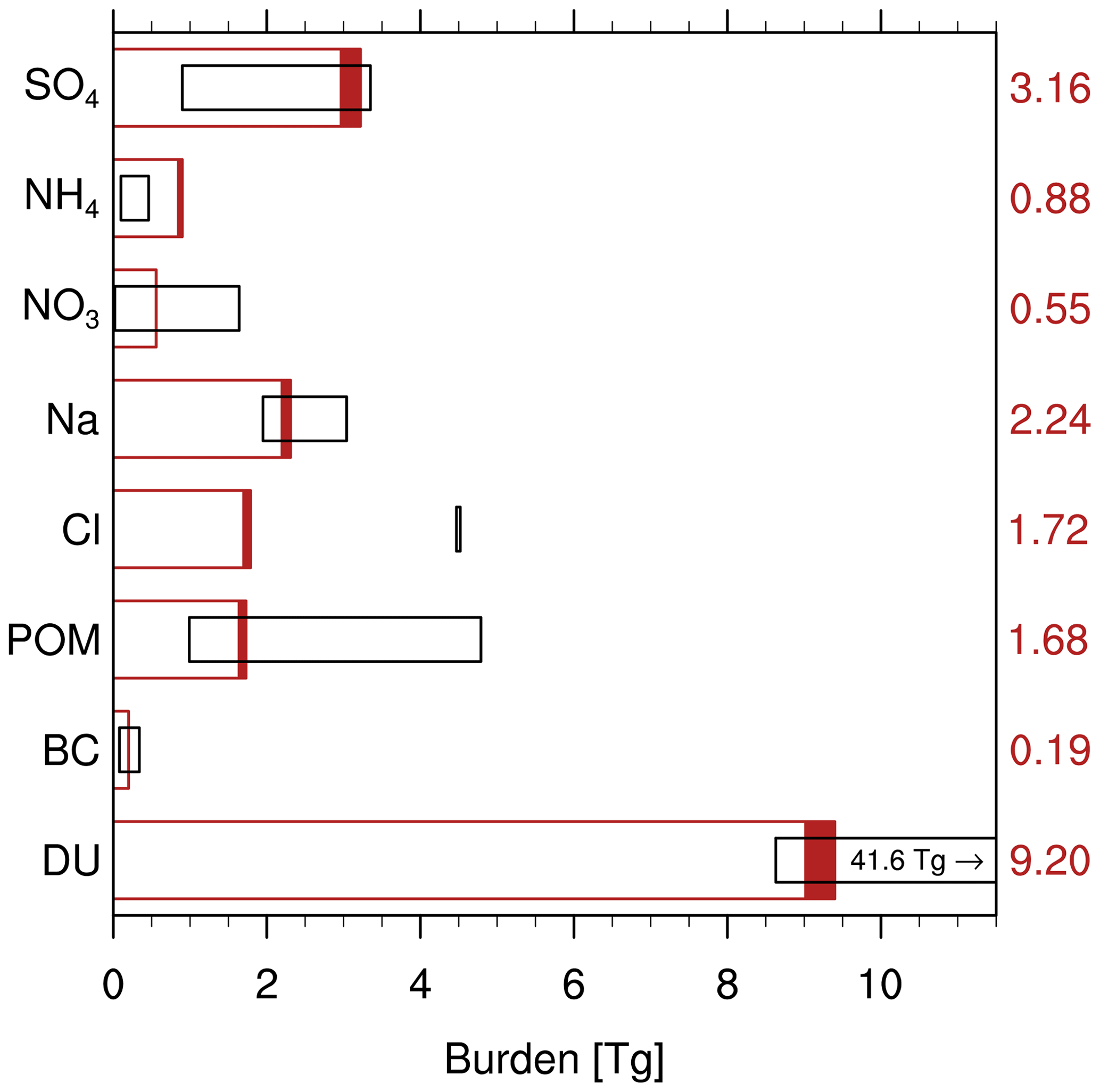
Figure 11Global tropospheric aerosol burdens simulated by EMAC (MADE3). The filled part of each bar indicates the range of values for the 10 evaluated years. The average values are given on the right side of the plot. The ranges of values from a compilation of other studies (Lauer and Hendricks, 2006; Textor et al., 2006; Bauer et al., 2008; Wang et al., 2009; Mann et al., 2010; Pozzer et al., 2012; Kirkevåg et al., 2013; He and Zhang, 2014; van Noije et al., 2014; Lee et al., 2015; Michou et al., 2015) are shown as black boxes. Values for NH4 have only been reported by Lauer and Hendricks (2006), Pozzer et al. (2012), and He and Zhang (2014); values for Cl only by He and Zhang (2014). The reported literature range for Na is for sea spray. Note that this is not meant to be a comprehensive literature review but should yield a representative picture. Extreme values may stem from specific sensitivity studies.
The species' burdens simulated with MADE3 in EMAC mostly fall within the ranges of previous estimates. For the secondary inorganic species, i.e., SO4, NH4, and NO3, the partitioning between the coarse and fine modes appears to play an important role. While SO4 and NH4 are found predominantly in the fine modes (>95 % on average), NO3 partitions roughly equally between the fine and coarse modes on average. While the SO4 and NH4 burdens simulated in the present study are at the upper end of the range of available model results, the NO3 burden lies well within the range spanned by the other studies. This could be an indication that especially the lifetimes of fine-mode aerosol are comparatively long in our model. The comparatively high NH4 burden in our simulation should be interpreted with care, since corresponding values have been reported by only three other studies.
The primary species' residence times estimated from our simulation amount to 8.84, 8.92, and 2.00 days for BC, POM, and DU, respectively. These values fall in the ranges of [2.44, 9.60], [2.56, 9.52], and [1.56, 5.92] days, respectively, spanned by the results of the previous studies mentioned in the caption of Fig. 11.
We implemented the aerosol microphysics submodel MADE3 into the global chemistry–climate model EMAC as a successor to MADE and MADE-in. The new submodel version includes nine aerosol species and represents three types of aerosol particles in three different size ranges. With respect to its predecessors, MADE3 now explicitly simulates the partitioning between the gas and the aerosol phase in the coarse mode, as well as the interactions between the coarse and fine modes, and includes a fully revised coupling to the scavenging submodel accounting for the wet deposition processes.
As a first application, we performed a 10-year model simulation. To evaluate the model quality, we compared the simulation output to data from a wide range of observations. These include aerosol (species) mass and number concentrations, size distributions, and AOD from surface-based, airborne, and satellite measurements. The results of these comparisons are summarized below.
The main conclusion from the near-surface mass concentration comparisons is that EMAC with MADE3 mostly captures the observed annual average spatial patterns of all aerosol species included in the model. Best agreement was obtained for black carbon and particulate organic matter, but they could only be compared over the US. Among the other species, quantitative agreement is typically best for SO4, with up to ∼70 % of the simulated monthly mean values within a factor of 2 of the observations (factors between 0.5 and 2). Concentrations of the nitrogen-containing components, NH4 and NO3, are spatially less heterogeneous in the simulations than in the observations. This is likely caused by the coarse model resolution and by higher temporal variability of the precursor emissions compared to those of SO4, which leads to larger uncertainties in the emission datasets. We detected a high bias of the average of the simulated values vs. the observations for nearly all species, which might have to do with underestimated removal of fine-mode particles from the atmosphere. Note, however, that near-surface mass concentrations could only be evaluated over the Northern Hemisphere continents, with very few exceptions. The comparisons demonstrate the ability of EMAC with MADE3 to simulate several tens of percent of the monthly mean aerosol species concentrations within a factor of 2 of the observations, which indicates a quality of the model that is similar to that of other global aerosol models.
The comparison of vertical BC mass mixing ratio and aerosol particle number concentration profiles revealed that the model representations of aerosol vertical transport and wet removal may need to be improved in order to avoid overestimations of the upper tropospheric aerosol load. In addition, the model description of new particle formation needs to be further developed towards more robust representations of particle formation from inorganic and also organic aerosol precursors. In this context, it should be stressed again that discrepancies in the representation of vertical aerosol profiles are a common feature of current global aerosol models and need to be the subject of in-depth investigations and resulting model improvements in the future.
Simulated near-surface size distributions, or rather their level of agreement with observations, were strongly affected by the coarse spatial and temporal model resolution. The simulated distributions agreed well with measurements in areas representative of continental background conditions. However, a rather unimodal shape could often be seen in the simulation, whereas observed distributions contained a separate nucleation mode in many cases, for instance. This could be due to the coarse model resolution which impedes the representation of local enhancements of ultrafine particles due to local emissions. It could also be a consequence of the relatively wide MADE3 modes in comparison to those fitted to the observational data. Furthermore, weaker seasonal variability was found in the simulation than in observations across Europe. For the future, we plan to deepen this analysis by extending the simulated period and include comparisons to the data collected by Asmi et al. (2011) and Birmili et al. (2016). This should also include simulations with alternative assumptions for the mode widths and with a higher spatial resolution.
The comparison of simulated AOD to ground-based and satellite observations provided further evidence for some of the conclusions drawn above. Compared to the observational data, our model shows larger AOD in regions affected by anthropogenic pollution and biomass burning emissions. In contrast, the simulated AOD is smaller compared to the observations over regions where DU dominates the aerosol composition. Together with deviations in sea-spray-dominated areas, this shows the necessity of improving the representation of wind-driven dust and sea spray emissions in the model.
Our evaluation also included a comparison with electron microscopy measurements, suggesting that the model is largely able to simulate the dependence of aerosol composition on particle size. However, these analyses need to be extended in the future to draw more robust conclusions. Simulations of specific episodes during which measurements were taken are required for this purpose, with the appropriate meteorology and emissions. The size distribution of the measured aerosol particles should also be taken into account for a thorough comparison, especially that of the surface area available for SO4 condensation, in order to understand the model–observation differences in the representation of fine-mode sulfate in more detail. Such data, however, have to be measured with different instruments. For some campaigns, both size-resolved composition and size distribution measurements are available. MADE3 can be evaluated with these data in the future by applying the method presented here.
We mentioned many sources of uncertainty in the parameters and parameterizations that are part of the aerosol microphysics and transport calculations, e.g., related to new particle formation and convective transport or scavenging. In addition, there are numerous issues that have to be taken into account when comparing simulations to observations. Among those are the specific meteorological conditions and emissions, which influenced the measured aerosol properties, the correspondence of measured and simulated species, and the uncertainties inherent in the observations, which are rarely reported. A detailed analysis of all these factors is beyond the scope of the present study. Our main conclusion here is that, in all “disciplines”, the simulation with MADE3 achieved a level of agreement with observations that falls within the range of results reported by other authors from simulations with their models. The same mostly holds for burdens and residence times of the MADE3 aerosol components, so that the new submodel can be considered ready for application.
Future studies with MADE3 should focus on the analysis and reduction of the model discrepancies highlighted in the present evaluation. This could include, for example, the consideration of observational uncertainties, a detailed analysis of the scavenging efficiency and its dependency on the aerosol size distributions and the underlying microphysical processes, as well as simulations with higher spatial resolution and model experiments focusing on the new particle formation processes considering different nucleation parameterizations.
One of the intended applications of MADE3 in EMAC is the reassessment of the aerosol-induced ship emissions effect on climate as described in the introduction. We saw much higher aerosol nitrate concentrations over the major shipping routes in our present simulation than in previous simulations with MADE, where interactions of the coarse mode with the gas phase were limited to the exchange of water in the condensed and gaseous phases. Hence, previous conclusions, especially in terms of the assessment of low-sulfur fuel scenarios, might have to be reconsidered.
Furthermore, MADE3 will be used as part of EMAC to assess climate effects of the aerosol through modification of ice and mixed-phase cloud properties. MADE3 is especially suitable for such applications due to its mixing state representation with fully soluble, mixed, and insoluble particles in each of the three size ranges of the Aitken, accumulation, and coarse modes.
MESSy is continuously further developed and applied by a consortium of institutions. The usage of MESSy, including MADE3, and access to the source code is licensed to all affiliates of institutions which are members of the MESSy Consortium. Institutions can become members of the MESSy Consortium by signing the MESSy Memorandum of Understanding. More information can be found on the MESSy Consortium website (http://www.messy-interface.org, last access: 9 January 2019). The model configuration discussed in this paper has been developed based on version 2.53 and will be part of the next EMAC release (version 2.54).
ESMValTool v1.0, used for evaluating EMAC (MADE3) in comparison with observations, is available at https://doi.org/10.17874/ac8548f0315 (see also http://www.esmvaltool.org, last access: 9 January 2019).
The model simulation data analyzed in this work are available at https://doi.org/10.1594/WDCC/EMAC-MADE3_monthly (Righi, 2018a) and https://doi.org/10.1594/WDCC/EMAC-MADE3_12h (Righi, 2018b).
| ACCESS | Arctic Climate Change, Economy, and Society project |
| ACCESS-2 | Alternative fuel effects on Contrails and Cruise EmiSSions 2 |
| AEROCOM | AEROsol Comparisons between Observations and Models |
| AERONET | AErosol RObotic NETwork |
| AGAGE | Advanced Global Atmospheric Gases Experiment |
| AOD | Aerosol optical depth |
| ATSR | Along-Track Scanning Radiometer |
| BC | Black carbon |
| CAM | Community Atmosphere Model |
| CASTNET | Clean Air Status and Trends NETwork |
| Cl | Chloride |
| CONCERT | CONtrail and Cirrus ExpeRimenT |
| CR-AVE | Costa Rica Aura Validation Experiment |
| DC3 | Deep Convective Clouds and Chemistry project |
| DMS | Dimethyl sulfide |
| DU | Mineral dust aerosol |
| EANET | Acid Deposition Monitoring Network in East Asia |
| ECHAM5 | Fifth-generation European Centre Hamburg model |
| ECMWF | European Centre for Medium-Range Weather Forecasts |
| EMEP | European Monitoring and Evaluation Programme |
| ESMValTool | Earth System Model eValuation Tool |
| EMAC | ECHAM/MESSy Atmospheric Chemistry model |
| ESACCI | European Space Agency Climate Change Initiative |
| GMXe | Global Modal-aerosol eXtension |
| HadGEM3 | Hadley Global Environmental Model 3 |
| HAM | Hamburg Aerosol Model |
| HIPPO | HIAPER Pole-to-Pole Observations |
| IMPACT | Integrated Massively Parallel Atmospheric Chemical Transport |
| IMPROVE | Interagency Monitoring of Protected Visual Environments network |
| INCA | INterhemispheric differences in Cirrus properties from Anthropogenic emissions |
| LACE | Lindenberg Aerosol Characterization Experiment |
| MADE | Modal Aerosol Dynamics model for Europe, adapted for global applications |
| MATRIX | Multiconfiguration Aerosol TRacker of mIXing state |
| MESSy | Modular Earth Submodel System |
| MODIS | MODerate resolution Imaging Spectroradiometer |
| Na | Sea spray components other than chloride (mainly sodium; Na) |
| NH4 | Aerosol ammonium |
| NO3 | Aerosol nitrate |
| NOAA/ESRL | National Oceanic and Atmospheric Administration/Earth System Research Laboratory |
| Oct-AVE | October 2004 Aura Validation Experiment |
| POM | Particulate organic matter |
| SALTRACE | Saharan Aerosol Long-range Transport and Aerosol-Cloud-Interaction Experiment |
| SAMUM-2 | SAharan Mineral dUst experiMent 2 |
| SO4 | Aerosol sulfate |
| SOA | Secondary organic aerosol |
| SP2 | Single Particle Soot Photometer |
| SS | Sea spray aerosol |
| TC4 | Tropical Chemistry, Cloud, and Climate Coupling |
| TOMAS | TwO-Moment Aerosol Sectional microphysics model |
| UCN | Ultrafine condensation nuclei |
| UKCA | United Kingdom Chemistry and Aerosols model |
The submodel SCAV has undergone numerous updates since its original publication (Tost et al., 2006a), e.g., concerning the ice phase as described by Tost et al. (2010). Here, we briefly summarize the most important parts of the algorithms as currently implemented for MADE3 aerosol, as not all of them have been documented in the literature so far.
B1 Aerosol scavenging in and below clouds
Four operators are applied in the following sequence:
-
ice nucleation scavenging,
-
liquid nucleation scavenging,
-
snow impaction scavenging, and
-
rain impaction scavenging.
In SCAV, “nucleation scavenging” refers to both the actual nucleation of cloud droplets or ice crystals, and the scavenging of aerosol by cloud particles due to the aerosol particles' Brownian motion. The latter is currently not included in many global aerosol models, although it may have a substantial impact on particle number concentration (Pierce et al., 2015). The term “impaction scavenging” summarizes the processes through which aerosol particles are taken up by precipitation, i.e., falling hydrometeors. These processes include Brownian motion of aerosol particles towards hydrometeors, impaction of hydrometeors upon aerosol particles, and interception of aerosol particles by hydrometeors. A scavenging rate, η, which is applied to the number and mass concentrations of aerosol particles in grid boxes with clouds and/or precipitation, is computed for each mode and each of the above operators. It represents the aerosol fraction of the respective mode incorporated into cloud or precipitation particles during a model time step Δt.
Constant scavenging rates are assumed for ice nucleation:
where T is absolute temperature.
Cloud droplet nucleation is taken into account via an empirical function (Tost et al., 2006a) that is applied to the hydrophilic particles, i.e., to those in the soluble and mixed MADE3 modes:
with the dimensionless number median diameter of the respective aerosol mode (in units µm, as for Dg). For Brownian motion, a semi-empirical formulation of the scavenging coefficient by Pruppacher and Klett (1997) is used, which leads to a scavenging rate of (Tost et al., 2006a)
Here, LWC is the cloud liquid water content (mass per unit volume), and rcld is the effective cloud droplet radius, which is set constant at m. The aerosol particle diffusivity Δp is computed as
with Boltzmann constant kB, atmospheric dynamic viscosity , and the modal median diameter Dg. The factor
where λair stands for the mean free path of air, is known as the “slip correction”. Combining the two scavenging rates (Eqs. B2–B3), one arrives at the total liquid nucleation scavenged fraction per unit time:
The negative term accounts for the fact that aerosol particles cannot be scavenged at the same time by both nucleation and Brownian motion.
Snow impaction scavenging is parameterized as
with the snow mass flux Fs per unit area and time. The rate coefficient ηsim is applied to particles of all MADE3 modes.
Finally, the rain impaction scavenging rate is assumed to be (Tost et al., 2006a)
where Fr is the rain mass flux per unit area and time. The terms are computed for six different values of the rain droplet radius rr,i (), namely for 0.1 mm, 0.2 mm, 0.5 mm, 1 mm, 2 mm, and 5 mm. Ei is the collision efficiency of aerosol particles of size Dg and rain droplets of size rr,i as parameterized by Slinn (1984), with weights Wi based on the rain droplet radii rr,i. Compared to measurements, Ei is likely underestimated for fine-mode aerosol particles, which is a problem of any theoretically derived formulation for this parameter (Wang et al., 2010). As for snow impaction scavenging, the rain impaction scavenging rate is also applied to all MADE3 modes.
B2 Aerosol release from clouds and precipitation
In the case of evaporation or sublimation of cloud particles or precipitating hydrometeors, aerosol residues are released. The following section describes the algorithm for assigning the residual aerosol number and mass to the respective MADE3 modes. The mode naming convention for this section is described in the caption of Table 2. Corresponding numbers are as follows: ks=1, km=2, ki=3, as=4, am=5, ai=6, cs=7, cm=8, and ci=9.
B2.1 Assignment of aerosol particle number concentrations
Let and be the number concentrations of aerosol particles from mode q that were activated to form cloud droplets or ice crystals, respectively. Before all other calculations, the insoluble ice nuclei number concentrations are assigned to the corresponding mixed modes (according to assumption 4 in Sect. 2.3) and the Aitken-mode cloud particle cores to the corresponding accumulation modes (according to assumption 5):
These operations are not fully compatible with the nucleation scavenging scheme of SCAV (Appendix B1) since the nucleation scavenging rates considered by SCAV include not only nucleation of cloud droplets or ice crystals but also Brownian motion scavenging within clouds. Separating the rates of the different processes for considering the pure nucleation rate in the assignment of cloud residues to the MADE3 modes would require fundamental and very extensive changes of the SCAV core algorithm, which would be far beyond the scope the present study. We therefore apply a simplified approach here: Brownian motion scavenging of interstitial aerosol in non-precipitating clouds is particularly important in the case of ultrafine aerosols (e.g., Seinfeld and Pandis, 1998). Hence, large Brownian motion scavenging efficiencies of non-precipitating hydrometeors (nucleation scavenging operation in SCAV) can be expected in particular for the soluble Aitken mode, due to the small particle sizes of nucleating aerosol particles included in this mode. The insoluble and mixed Aitken modes as well as the accumulation and coarse modes contain larger particles, which are rather subject to nucleation scavenging than Brownian motion scavenging. Hence, we assume that only and include major contributions of Brownian scavenging. Therefore, this might lead to overestimations of the number of aerosol particles served as droplet or ice nuclei. In order to avoid this, we neglect and in Eq. (B9a) and (B9b), respectively. Due to its very small particle size, the soluble Aitken mode is only poorly activated to form cloud droplets and possible underestimations of due this simplification are probably small. In the case of ice scavenging, 5 % of Nks is assumed to serve as ice nuclei in the present version of SCAV (Sect. 2.3 and Appendix B1). At high concentrations of ultrafine soluble particles, this can lead to too-high values of compared to typical ice crystal concentrations in pristine cirrus clouds. Neglecting therefore has an additional benefit under these conditions. In the case of moderate number concentrations of the soluble Aitken mode, errors in the activated number caused by neglecting are probably small compared to the uncertainties inherent in the assumption of constant 5 % ice nucleation scavenging, since variations of ice crystal number concentrations resulting from varying homogeneous freezing conditions (e.g., Kärcher and Lohmann, 2002) cannot be represented. Sensitivity simulations showed, however, that neglecting and in Eq. (B9a) and (B9b) has only a marginal effects on the results. This means that the effect of Brownian motion scavenging is small and further demonstrates that possible errors due to the simplified representations of nucleation scavenging are limited. Nevertheless, separate budgeting of processes in the nucleation scavenging algorithm of SCAV could be the subject of future model development work to enable a fully consistent representation of cloud residual aerosol.
The number concentrations denoted by N will be the output values of the mode assignment algorithm and are further modified as described in the following. According to Eq. (B9a)–(B9h), cloud particle cores now belong to one of the four modes: q=4, 5, 7, and 8 (corresponding to “as”, “am”, “cs”, and “cm”). In the following, the symbol
is used to represent their cumulative number concentration in mode q. The total number concentration of cloud particles is assumed to remain constant, i.e.,
during the mode assignment process. In the present setup, SCAV later reduces all to 10 % of their values to account for coagulation of cloud particles.
Now, the numbers of cloud particle cores per unit volume that are transferred to different aerosol modes upon evaporation/sublimation of the cloud/precipitation particles are calculated. Such transfers are due to impaction scavenging of other aerosol particles. The symbol for the number of cores transferred from mode q to mode r per unit volume shall be ; the symbol for the total number of interstitial mode q particles per unit volume collected by impaction scavenging (Sect. B1) shall be .
Different cloud or precipitation particles can scavenge very different numbers of interstitial aerosol particles from the various aerosol modes leading to a large variety of modifications of the original cloud particle cores. Representative mathematical descriptions of this complex system would lead to overly complicated and error-prone formulas to describe mode transfers of cores upon evaporation/sublimation of the cloud or precipitation particles. Hence, the system is simplified here by
-
maximizing the transfer to mixed modes and
-
maximizing the transfer to coarse modes,
with a higher priority of the transfers to mixed modes. This deliberate “overestimation” of the transfer rates is motivated by the fact that the general reduction of (to 10 %; see above) does not account for transfers of cloud particle cores to other modes, since it simply reduces the total number of residues. The simplifications imply, for instance, the assumption that the interactions of interstitial aerosol with cloud particles are as homogeneously distributed as possible over the cloud particle population, maximizing the number of transfers induced by impaction scavenging.
It is acknowledged that these simplifications are somewhat arbitrary, but judging by previous simulation results – e.g., a relatively small long-term mean effect of cloud processing on aerosol particle aging when compared to condensation of trace gases – the associated error is expected to be tolerable. Nevertheless, different assumptions should be tested in the future in order to explore the sensitivity of the simulation results to different representations of the mode transfers.
No transfer is required out of mode “cm”, as this mode represents the highest degree of aerosol particle mixing and aging. For modes “cs” and “am”, the calculation is straightforward, as cores from these modes can only be transferred to mode “cm” (or remain in their respective mode). With the simplifying assumptions described above, we obtain
Here,
is the fraction that mode q contributes to the total number concentration of cloud particle cores. The MIN operation is required because cloud or precipitation particles can collect multiple other particles via impaction scavenging, but their cores can of course only be transferred to mode “cm” once.
The situation is more complicated for the “as” cores, as they can be transferred to modes “am”, “cs”, and “cm”, depending on the aerosol particles taken up by impaction scavenging. To simplify the system by maximizing the transfer to the mixed and/or coarse modes, the following assumptions are made:
-
collected aerosol particles that contain insoluble material are distributed as evenly as possible over the “as” cores;
-
fine, i.e., Aitken- and accumulation-mode, particles that contain insoluble material are collected preferentially by cloud or precipitation particles that have not collected coarse-mode particles which contain insoluble material; and
-
soluble coarse-mode particles are collected preferentially by cloud or precipitation particles that have also collected fine-mode particles that contain insoluble material.
Figure B1 may help the reader visualize these assumptions and the associated transfers described in the following.
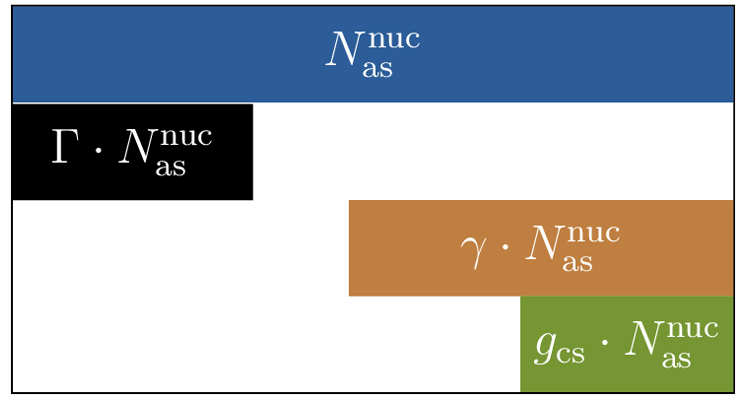
Figure B1Illustration of the transfer concept for mode “as” cloud particle cores upon impaction scavenging of particles that induce such transfers. The blue bar represents the number concentration of mode “as” aerosol particles that have nucleated cloud particles; the boxes below it represent the number concentrations of impaction-scavenged aerosol particles from the three considered classes: coarse-mode particles that contain insoluble material (black), fine-mode particles that contain insoluble material (brown), and soluble coarse-mode particles (green). See text for explanation of the symbols.
Following the outlined scheme, the direct transfer of cloud or precipitation particle cores from mode “as” to mode “cm” by impaction of aerosol particles from modes “cm” and “ci” is considered first. Let
be the fraction of mode r cores that has collected aerosol particles from the coarse modes that contain insoluble material (which is actually independent of r). Here,
is the fraction of cores from mode r (or, in fact, from any mode) that has collected aerosol particles from mode q. The number of cores directly transferred from mode “as” to mode “cm” per unit volume can then be written as
Subsequently, if any “as” cores remain, transfers from mode “as” to mode “cm” via collection of both fine-mode particles that contain insoluble material and soluble coarse-mode particles are considered. Let
be the analogue to Γ (Eq. B14) for collected fine-mode particles that contain insoluble material. The expression for the transfer of “as” cores to “cm” via collection of (at least) two aerosol particles (one from “km”, “ki”, “am”, or “ai” and one from “cs”) per cloud/precipitation particle then reads
Once the terms for transfer to the mixed coarse mode have been established, and in case any “as” cores remain, transfer from “as” to “am” or “cs” without subsequent transfer to “cm” also has to be considered:
After these calculations, the values are redistributed accordingly (read the arrows as “new value on the left-hand side is computed from old values on the right-hand side”):
B2.2 Assignment of aerosol particle mass concentrations
As for the number concentrations, let and be the mass concentrations of aerosol particles from mode q that were activated to form cloud droplets or ice crystals, respectively. Furthermore, let be the aerosol mass per unit volume generated within, or lost from, the cloud particles nucleated by mode q aerosol particles, which is due to cloud liquid-phase chemistry.
For consistency with the number treatment, the insoluble ice nuclei mass concentrations are first assigned to the corresponding mixed modes and the Aitken-mode cloud particle cores to the corresponding accumulation modes:
Note that, in contrast to the treatment of particle numbers (Eq. B9a and B9b), and need to be considered here, since Brownian motion scavenging contributes to the mass of cloud particle residues while their number concentration does not change. As before, the m will be the output values of our algorithm; the 𝔪 values are the input values, computed by SCAV using the fractions given in Appendix B1. Note that there are no () values because ice-phase chemistry is not considered.
In order to simplify the following expressions, the cloud particle core mass concentration for each mode (“as”, “am”, “cs”, and “cm”) is defined as the sum of the activated mode r aerosol particle mass per unit volume, the aerosol mass generated per unit volume within or on the cloud particles nucleated by mode r aerosol particles or lost from these cloud particles, and the mass concentration of collected aerosol particles that do not induce transfers of the residual from mode r (let be the total mass concentration of mode q particles that are collected by impaction scavenging; Appendix B1):
Now, the transferred mass concentrations are computed. For each mode (q=4, 5, 7, and 8), the mass concentrations of particles that induced the transfer and a fraction of the core mass concentration have to be transferred. As mode “cm” is only a target mode for residuals, there is no transfer out of this mode. All the mass that it receives by impaction scavenging stays in mode “cm”. For the other three modes of cloud particle cores (“cs”, “am”, “as”), the mass concentrations that have to be transferred are calculated consistently with the number transfers:
Notes:
-
This term denotes the fraction of collected fine-mode particles containing insoluble material that is transferred with .
-
This term denotes the fraction of collected soluble coarse-mode particles that is transferred with .
Similar notes apply to the first terms in Eqs. (B27)–(B29). Note also that the terms with denominator add up to 1, and the terms with denominator do so as well. This is due to the limits imposed on the number transfers (see Sect. B2.1).
Finally, the mass concentration assignments are performed in an analogous manner as is done for the numbers (again, read the arrows as “new value on the left-hand side is computed from old values on the right-hand side”):
In the model, these expressions are applied to the individual species mass concentrations since they are the central prognostic quantities. This is legitimate since the individual species redistribute in proportion to the total mass.
The supplement related to this article is available online at: https://doi.org/10.5194/gmd-12-541-2019-supplement.
JCK, with the aid of JH and MR, implemented the aerosol submodel MADE3 into the EMAC model system, performed the reference simulation and its evaluation, and wrote the paper. PJ helped to implement MADE3 and to prepare the simulation setup. HT assisted in the coupling of MADE3 to the EMAC scavenging submodel SCAV. KK provided the electron microscopy data and helped in the comparison with the model results. BW, DS, KH, JPS, and AEP provided data from aircraft-based observations and assisted in the corresponding model evaluation. TP provided the ESACCI data and contributed to the comparison of the simulations with the satellite-based observations. All coauthors assisted in the preparation of the paper.
The authors declare that they have no conflict of interest.
This study was supported by the DLR transport program (projects “Global
model studies on the effects of transport-induced aerosols on ice clouds and
climate” and “Transport and the Environment – VEU2”). Additional support
was received from the DLR space research program (project “Climate relevant
trace gases, aerosols and clouds – KliSAW”). The EMAC simulations were
performed at the German Climate Computing Center (DKRZ, Hamburg, Germany),
which also provided kind support for long-term storage of the model output
analyzed in this work. The DLR SP2 data were obtained with the support of the
Helmholtz Association under grant no. VH-NG-606
(Helmholtz-Hochschul-Nachwuchsforschergruppe AerCARE), the European Union
project ACCESS under grant agreement no. 265863, and DLR projects VolcATS and
CATS. The analysis of the DLR SP2 data was supported in part by the Center
for Advanced Studies at LMU, LMU Munich's Institutional Strategy LMUexcellent
within the framework of the German Excellence Initiative, and by the European
Research Council under the European Community's Horizon 2020 research and
innovation framework program/ERC grant agreement no. 640458 – A-LIFE. The
ESACCI data were provided by the ESA Climate Change Initiative and in
particular its Aerosol_cci project (http://www.esa-aerosol-cci.org,
last access: 9 January 2019) from the individual provider Swansea University
(Peter North). Mattia Righi received funding from the Initiative and
Networking Fund of the Helmholtz Association through the project “Advanced
Earth System Modelling Capacity (ESM)”. Konrad Kandler acknowledges support by the Deutsche
Forschungsgemeinschaft (DFG grants KA 2280/2 and /3). We are grateful to
Phoebe Graf (DLR) for processing the greenhouse gas data used in the TNUDGE
submodel, and to Mariano Mertens and Christof Beer
(DLR) as well as two anonymous
referees for their valuable comments on the manuscript. Last but not least,
we thank the whole MESSy team (developers and maintainers) for the invaluable
support.
The article processing charges for this open-access
publication were covered by a Research
Centre of the
Helmholtz Association.
Edited by: Samuel Remy
Reviewed by: two anonymous referees
Allen, R. J. and Landuyt, W.: The vertical distribution of black carbon in CMIP5 models: Comparison to observations and the importance of convective transport, J. Geophys. Res.-Atmos., 119, 4808–4835, https://doi.org/10.1002/2014JD021595, 2014. a, b
AMEC Environment & Infrastructure, I.: Clean Air Status and Trends Network (CASTNET) 2013 Annual Report, Tech. rep., U.S. Environmental Protection Agency, Washington, DC, USA, available at: http://epa.gov/castnet/javaweb/docs/annual_report_2013.pdf (last access: 9 January 2019), 2015. a
Ames, R. B. and Malm, W. C.: Comparison of sulfate and nitrate particle mass concentrations measured by IMPROVE and the CDN, Atmos. Environ., 35, 905–916, https://doi.org/10.1016/S1352-2310(00)00369-1, 2001. a, b
Ansmann, A., Petzold, A., Kandler, K., Tegen, I., Wendisch, M., Müller, D., Weinzierl, B., Müller, T., and Heintzenberg, J.: Saharan Mineral Dust Experiments SAMUM-1 and SAMUM-2: what have we learned?, Tellus B, 63, 403–429, https://doi.org/10.1111/j.1600-0889.2011.00555.x, 2011. a
Aquila, V.: Global model studies on the distribution and composition of potential atmospheric ice nuclei, Ph.D. thesis, LMU München, Germany, available at: http://elib.dlr.de/61778/1/Aquila-diss-FINAL-20091215.pdf (last access: 9 January 2019), 2009. a
Aquila, V., Hendricks, J., Lauer, A., Riemer, N., Vogel, H., Baumgardner, D., Minikin, A., Petzold, A., Schwarz, J. P., Spackman, J. R., Weinzierl, B., Righi, M., and Dall'Amico, M.: MADE-in: a new aerosol microphysics submodel for global simulation of insoluble particles and their mixing state, Geosci. Model Dev., 4, 325–355, https://doi.org/10.5194/gmd-4-325-2011, 2011. a, b, c, d, e, f, g
Asmi, A., Wiedensohler, A., Laj, P., Fjaeraa, A.-M., Sellegri, K., Birmili, W., Weingartner, E., Baltensperger, U., Zdimal, V., Zikova, N., Putaud, J.-P., Marinoni, A., Tunved, P., Hansson, H.-C., Fiebig, M., Kivekäs, N., Lihavainen, H., Asmi, E., Ulevicius, V., Aalto, P. P., Swietlicki, E., Kristensson, A., Mihalopoulos, N., Kalivitis, N., Kalapov, I., Kiss, G., de Leeuw, G., Henzing, B., Harrison, R. M., Beddows, D., O'Dowd, C., Jennings, S. G., Flentje, H., Weinhold, K., Meinhardt, F., Ries, L., and Kulmala, M.: Number size distributions and seasonality of submicron particles in Europe 2008-2009, Atmos. Chem. Phys., 11, 5505–5538, https://doi.org/10.5194/acp-11-5505-2011, 2011. a
Bauer, S. E., Wright, D. L., Koch, D., Lewis, E. R., McGraw, R., Chang, L.-S., Schwartz, S. E., and Ruedy, R.: MATRIX (Multiconfiguration Aerosol TRacker of mIXing state): an aerosol microphysical module for global atmospheric models, Atmos. Chem. Phys., 8, 6003–6035, https://doi.org/10.5194/acp-8-6003-2008, 2008. a, b, c
Bellouin, N., Rae, J., Jones, A., Johnson, C., Haywood, J., and Boucher, O.: Aerosol forcing in the Climate Model Intercomparison Project (CMIP5) simulations by HadGEM2-ES and the role of ammonium nitrate, J. Geophys. Res.-Atmos., 116, D20206, https://doi.org/10.1029/2011JD016074, 2011. a
Bevan, S. L., North, P. R. J., Los, S. O., and Grey, W. M. F.: A global dataset of atmospheric aerosol optical depth and surface reflectance from AATSR, Remote Sens. Environ., 116, 199–210, https://doi.org/10.1016/j.rse.2011.05.024, 2012. a
Birmili, W., Alaviippola, B., Hinneburg, D., Knoth, O., Tuch, T., Borken-Kleefeld, J., and Schacht, A.: Dispersion of traffic-related exhaust particles near the Berlin urban motorway – estimation of fleet emission factors, Atmos. Chem. Phys., 9, 2355–2374, https://doi.org/10.5194/acp-9-2355-2009, 2009. a, b
Birmili, W., Weinhold, K., Rasch, F., Sonntag, A., Sun, J., Merkel, M., Wiedensohler, A., Bastian, S., Schladitz, A., Löschau, G., Cyrys, J., Pitz, M., Gu, J., Kusch, T., Flentje, H., Quass, U., Kaminski, H., Kuhlbusch, T. A. J., Meinhardt, F., Schwerin, A., Bath, O., Ries, L., Gerwig, H., Wirtz, K., and Fiebig, M.: Long-term observations of tropospheric particle number size distributions and equivalent black carbon mass concentrations in the German Ultrafine Aerosol Network (GUAN), Earth Syst. Sci. Data, 8, 355–382, https://doi.org/10.5194/essd-8-355-2016, 2016. a
Bond, T. C. and Bergstrom, R. W.: Light absorption by carbonaceous particles: An investigative review, Aerosol Sci. Tech., 40, 27–67, https://doi.org/10.1080/02786820500421521, 2006. a
Boulon, J., Sellegri, K., Hervo, M., Picard, D., Pichon, J.-M., Fréville, P., and Laj, P.: Investigation of nucleation events vertical extent: a long term study at two different altitude sites, Atmos. Chem. Phys., 11, 5625–5639, https://doi.org/10.5194/acp-11-5625-2011, 2011. a
Brinkop, S. and Sausen, R.: A Finite Difference Approximation for Convective Transports which Maintains Positive Tracer Concentrations, Beitr. Physik Atmosphäre, 70, 245–248, 1997. a
Capaldo, K., Corbett, J. J., Kasibhatla, P., Fischbeck, P., and Pandis, S. N.: Effects of ship emissions on sulphur cycling and radiative climate forcing over the ocean, Nature, 400, 743–746, 1999. a
Carslaw, K. S., Lee, L. A., Reddington, C. L., Pringle, K. J., Rap, A., Forster, P. M., Mann, G. W., Spracklen, D. V., Woodhouse, M. T., Regayre, L. A., and Pierce, J. R.: Large contribution of natural aerosols to uncertainty in indirect forcing, Nature, 503, 67–71, https://doi.org/10.1038/nature12674, 2013. a
Carslaw, K. S., Gordon, H., Hamilton, D. S., Johnson, J. S., Regayre, L. A., Yoshioka, M., and Pringle, K. J.: Aerosols in the Pre-industrial Atmosphere, Curr. Clim. Change Rep., 3, 1–15, https://doi.org/10.1007/s40641-017-0061-2, 2017. a
Cavalli, F., Facchini, M. C., Decesari, S., Mircea, M., Emblico, L., Fuzzi, S., Ceburnis, D., Yoon, Y. J., O'Dowd, C. D., Putaud, J.-P., and Dell'Acqua, A.: Advances in characterization of size-resolved organic matter in marine aerosol over the North Atlantic, J. Geophys. Res.-Atmos., 109, D24215, https://doi.org/10.1029/2004JD005137, 2004. a
Chandra, I., Kim, S., Seto, T., Otani, Y., Takami, A., Yoshino, A., Irei, S., Park, K., Takamura, T., Kaneyasu, N., and Hatakeyama, S.: New particle formation under the influence of the long-range transport of air pollutants in East Asia, Atmos. Environ., 141, 30–40, https://doi.org/10.1016/j.atmosenv.2016.06.040, 2016. a
Clarke, A. D. and Kapustin, V. N.: A Pacific Aerosol Survey. Part I: A Decade of Data on Particle Production, Transport, Evolution, and Mixing in the Troposphere, J. Atmos. Sci., 59, 363–382, https://doi.org/10.1175/1520-0469(2002)059<0363:APASPI>2.0.CO;2, 2002. a, b
Cooke, W. F., Liousse, C., Cachier, H., and Feichter, J.: Construction of a 1∘ × 1∘ fossil fuel emission data set for carbonaceous aerosol and implementation and radiative impact in the ECHAM4 model, J. Geophys. Res.-Atmos., 104, 22137–22162, https://doi.org/10.1029/1999JD900187, 1999. a
Corbett, J. J., Winebrake, J. J., Green, E. H., Kasibhatla, P., Eyring, V., and Lauer, A.: Mortality from Ship Emissions: A Global Assessment, Environ. Sci. Tech., 41, 8512–8518, https://doi.org/10.1021/es071686z, 2007. a
Croft, B., Pierce, J. R., Martin, R. V., Hoose, C., and Lohmann, U.: Uncertainty associated with convective wet removal of entrained aerosols in a global climate model, Atmos. Chem. Phys., 12, 10725–10748, https://doi.org/10.5194/acp-12-10725-2012, 2012. a
Dahlkötter, F., Gysel, M., Sauer, D., Minikin, A., Baumann, R., Seifert, P., Ansmann, A., Fromm, M., Voigt, C., and Weinzierl, B.: The Pagami Creek smoke plume after long-range transport to the upper troposphere over Europe – aerosol properties and black carbon mixing state, Atmos. Chem. Phys., 14, 6111–6137, https://doi.org/10.5194/acp-14-6111-2014, 2014. a
de Leeuw, G., Holzer-Popp, T., Bevan, S., Davies, W. H., Descloitres, J., Grainger, R. G., Griesfeller, J., Heckel, A., Kinne, S., Klüser, L., Kolmonen, P., Litvinov, P., Martynenko, D., North, P., Ovigneur, B., Pascal, N., Poulsen, C., Ramon, D., Schulz, M., Siddans, R., Sogacheva, L., Tanré, D., Thomas, G. E., Virtanen, T. H., von Hoyningen Huene, W., Vountas, M., and Pinnock, S.: Evaluation of seven European aerosol optical depth retrieval algorithms for climate analysis, Rem. Sens. Environ., 162, 295–315, https://doi.org/10.1016/j.rse.2013.04.023, 2015. a
Dentener, F., Kinne, S., Bond, T., Boucher, O., Cofala, J., Generoso, S., Ginoux, P., Gong, S., Hoelzemann, J. J., Ito, A., Marelli, L., Penner, J. E., Putaud, J.-P., Textor, C., Schulz, M., van der Werf, G. R., and Wilson, J.: Emissions of primary aerosol and precursor gases in the years 2000 and 1750 prescribed data-sets for AeroCom, Atmos. Chem. Phys., 6, 4321–4344, https://doi.org/10.5194/acp-6-4321-2006, 2006. a, b, c, d, e, f, g, h
Dietmüller, S., Jöckel, P., Tost, H., Kunze, M., Gellhorn, C., Brinkop, S., Frömming, C., Ponater, M., Steil, B., Lauer, A., and Hendricks, J.: A new radiation infrastructure for the Modular Earth Submodel System (MESSy, based on version 2.51), Geosci. Model Dev., 9, 2209–2222, https://doi.org/10.5194/gmd-9-2209-2016, 2016. a, b, c, d, e
Eyring, V., Righi, M., Lauer, A., Evaldsson, M., Wenzel, S., Jones, C., Anav, A., Andrews, O., Cionni, I., Davin, E. L., Deser, C., Ehbrecht, C., Friedlingstein, P., Gleckler, P., Gottschaldt, K.-D., Hagemann, S., Juckes, M., Kindermann, S., Krasting, J., Kunert, D., Levine, R., Loew, A., Mäkelä, J., Martin, G., Mason, E., Phillips, A. S., Read, S., Rio, C., Roehrig, R., Senftleben, D., Sterl, A., van Ulft, L. H., Walton, J., Wang, S., and Williams, K. D.: ESMValTool (v1.0) – a community diagnostic and performance metrics tool for routine evaluation of Earth system models in CMIP, Geosci. Model Dev., 9, 1747–1802, https://doi.org/10.5194/gmd-9-1747-2016, 2016. a
Gao, W., Fan, J., Easter, R. C., Yang, Q., Zhao, C., and Ghan, S. J.: Coupling spectral-bin cloud microphysics with the MOSAIC aerosol model in WRF-Chem: Methodology and results for marine stratocumulus clouds, J. Adv. Model Earth Syst., 8, 1289–1309, https://doi.org/10.1002/2016MS000676, 2016. a
García, M. I., Rodríguez, S., González, Y., and García, R. D.: Climatology of new particle formation at Izaña mountain GAW observatory in the subtropical North Atlantic, Atmos. Chem. Phys., 14, 3865–3881, https://doi.org/10.5194/acp-14-3865-2014, 2014. a
Gettelman, A., Liu, X., Barahona, D., Lohmann, U., and Chen, C.: Climate impacts of ice nucleation, J. Geophys. Res.-Atmos., 117, d20201, https://doi.org/10.1029/2012JD017950, 2012. a
Giamarelou, M., Eleftheriadis, K., Nyeki, S., Tunved, P., Torseth, K., and Biskos, G.: Indirect evidence of the composition of nucleation mode atmospheric particles in the high Arctic, J. Geophys. Res.-Atmos., 121, 965–975, https://doi.org/10.1002/2015JD023646, 2016. a
Guelle, W., Schulz, M., Balkanski, Y., and Dentener, F.: Influence of the source formulation on modeling the atmospheric global distribution of sea salt aerosol, J. Geophys. Res.-Atmos., 106, 27509–27524, https://doi.org/10.1029/2001JD900249, 2001. a, b
Guenther, A., Hewitt, C. N., Erickson, D., Fall, R., Geron, C., Graedel, T., Harley, P., Klinger, L., Lerdau, M., Mckay, W. A., Pierce, T., B, S., Steinbrecher, R., Tallamraju, R., Taylor, J., and Zimmerman, P.: A global model of natural volatile organic compound emissions, J. Geophys. Res., 100, 8873–8892, https://doi.org/10.1029/94JD02950, 1995. a
Hand, J. L., Copeland, S. A., Day, D. E., Dillner, A. M., Indresand, H., Malm, W. C., McDade, C. E., Moore, Jr., C. T. T., Pitchford, M. L., Schichtel, B. A., and Watson, J. G.: Spatial and Seasonal Patterns and Temporal Variability of Haze and its Constituents in the United States – Report V, Tech. rep., Cooperative Institute for Research in the Atmosphere, Colorado State University, Fort Collins, CO, USA, available at: http://vista.cira.colostate.edu/improve/Publications/Reports/2011/PDF/IMPROVE_V_FullReport.pdf (last access: 9 January 2019), 2011. a
Hara, K., Osada, K., Hayashi, M., Matsunaga, K., Shibata, T., Iwasaka, Y., and Furuya, K.: Fractionation of inorganic nitrates in winter Arctic troposphere: Coarse aerosol particles containing inorganic nitrates, J. Geophys. Res.-Atmos., 104, 23671–23679, https://doi.org/10.1029/1999JD900348, 1999. a
Hara, K., Nakazawa, F., Fujita, S., Fukui, K., Enomoto, H., and Sugiyama, S.: Horizontal distributions of aerosol constituents and their mixing states in Antarctica during the JASE traverse, Atmos. Chem. Phys., 14, 10211–10230, https://doi.org/10.5194/acp-14-10211-2014, 2014. a
He, J. and Zhang, Y.: Improvement and further development in CESM/CAM5: gas-phase chemistry and inorganic aerosol treatments, Atmos. Chem. Phys., 14, 9171–9200, https://doi.org/10.5194/acp-14-9171-2014, 2014. a, b, c
Hendricks, J., Kärcher, B., Lohmann, U., and Ponater, M.: Do aircraft black carbon emissions affect cirrus clouds on the global scale?, Geophys. Res. Lett., 32, l12814, https://doi.org/10.1029/2005GL022740, 2005. a
Hendricks, J., Kärcher, B., and Lohmann, U.: Effects of ice nuclei on cirrus clouds in a global climate model, J. Geophys. Res.-Atmos., 116, d18206, https://doi.org/10.1029/2010JD015302, 2011. a
Hjellbrekke, A.-G.: Data Report 2012 – Acidifying and eutrophying compounds and particulate matter, Tech. Rep. EMEP/CCC-Report 3/2014, Norwegian Institute for Air Research, Kjeller, Norway, available at: http://www.nilu.no/projects/ccc/reports/cccr3-2014.pdf (last access: 9 January 2019), 2014. a
Holben, B. N., Eck, T. F., Slutsker, I., Tanré, D., Buis, J. P., Setzer, A., Vermote, E., Reagan, J. A., Kaufman, Y. J., Nakajima, T., Lavenu, F., Jankowiak, I., and Smirnov, A.: AERONET–A Federated Instrument Network and Data Archive for Aerosol Characterization, Remote Sens. Environ., 66, 1–16, https://doi.org/10.1016/S0034-4257(98)00031-5, 1998. a
Holben, B. N., Tanré, D., Smirnov, A., Eck, T. F., Slutsker, I., Abuhassan, N., Newcomb, W. W., Schafer, J. S., Chatenet, B., Lavenu, F., Kaufman, Y. J., Castle, J. V., Setzer, A., Markham, B., Clark, D., Frouin, R., Halthore, R., Karneli, A., O'Neill, N. T., Pietras, C., Pinker, R. T., Voss, K., and Zibordi, G.: An emerging ground-based aerosol climatology: Aerosol optical depth from AERONET, J. Geophys. Res.-Atmos., 106, 12067–12097, https://doi.org/10.1029/2001JD900014, 2001. a
Holzer-Popp, T., de Leeuw, G., Griesfeller, J., Martynenko, D., Klüser, L., Bevan, S., Davies, W., Ducos, F., Deuzé, J. L., Graigner, R. G., Heckel, A., von Hoyningen-Hüne, W., Kolmonen, P., Litvinov, P., North, P., Poulsen, C. A., Ramon, D., Siddans, R., Sogacheva, L., Tanre, D., Thomas, G. E., Vountas, M., Descloitres, J., Griesfeller, J., Kinne, S., Schulz, M., and Pinnock, S.: Aerosol retrieval experiments in the ESA Aerosol_cci project, Atmos. Meas. Tech., 6, 1919–1957, https://doi.org/10.5194/amt-6-1919-2013, 2013. a
Hoose, C. and Möhler, O.: Heterogeneous ice nucleation on atmospheric aerosols: a review of results from laboratory experiments, Atmos. Chem. Phys., 12, 9817–9854, https://doi.org/10.5194/acp-12-9817-2012, 2012. a
Huneeus, N., Schulz, M., Balkanski, Y., Griesfeller, J., Prospero, J., Kinne, S., Bauer, S., Boucher, O., Chin, M., Dentener, F., Diehl, T., Easter, R., Fillmore, D., Ghan, S., Ginoux, P., Grini, A., Horowitz, L., Koch, D., Krol, M. C., Landing, W., Liu, X., Mahowald, N., Miller, R., Morcrette, J.-J., Myhre, G., Penner, J., Perlwitz, J., Stier, P., Takemura, T., and Zender, C. S.: Global dust model intercomparison in AeroCom phase I, Atmos. Chem. Phys., 11, 7781–7816, https://doi.org/10.5194/acp-11-7781-2011, 2011. a, b
Huntrieser, H., Lichtenstern, M., Scheibe, M., Aufmhoff, H., Schlager, H., Pucik, T., Minikin, A., Weinzierl, B., Heimerl, K., Pollack, I. B., Peischl, J., Ryerson, T. B., Weinheimer, A. J., Honomichl, S., Ridley, B. A., Biggerstaff, M. I., Betten, D. P., Hair, J. W., Butler, C. F., Schwartz, M. J., and Barth, M. C.: Injection of lightning-produced NOx, water vapor, wildfire emissions, and stratospheric air to the UT/LS as observed from DC3 measurements, J. Geophys. Res.-Atmos., 121, 6638–6668, https://doi.org/10.1002/2015JD024273, 2016. a
IMO: MARPOL Consolidated Edition 2011: Articles, Protocols, Annexes, Unified Interpretations of the International Convention for the Prevention of Pollution from Ships, 1973, as Modified by the 1978 and 1997 Protocols, IMO Publication, International Maritime Organization, 2011. a
IMPROVE: IMPROVE Data Guide, Tech. rep., University of California Davis, 1995. a
Jöckel, P., Tost, H., Pozzer, A., Brühl, C., Buchholz, J., Ganzeveld, L., Hoor, P., Kerkweg, A., Lawrence, M. G., Sander, R., Steil, B., Stiller, G., Tanarhte, M., Taraborrelli, D., van Aardenne, J., and Lelieveld, J.: The atmospheric chemistry general circulation model ECHAM5/MESSy1: consistent simulation of ozone from the surface to the mesosphere, Atmos. Chem. Phys., 6, 5067–5104, https://doi.org/10.5194/acp-6-5067-2006, 2006. a, b
Jöckel, P., Kerkweg, A., Pozzer, A., Sander, R., Tost, H., Riede, H., Baumgaertner, A., Gromov, S., and Kern, B.: Development cycle 2 of the Modular Earth Submodel System (MESSy2), Geosci. Model Dev., 3, 717–752, https://doi.org/10.5194/gmd-3-717-2010, 2010. a, b, c
Jöckel, P., Tost, H., Pozzer, A., Kunze, M., Kirner, O., Brenninkmeijer, C. A. M., Brinkop, S., Cai, D. S., Dyroff, C., Eckstein, J., Frank, F., Garny, H., Gottschaldt, K.-D., Graf, P., Grewe, V., Kerkweg, A., Kern, B., Matthes, S., Mertens, M., Meul, S., Neumaier, M., Nützel, M., Oberländer-Hayn, S., Ruhnke, R., Runde, T., Sander, R., Scharffe, D., and Zahn, A.: Earth System Chemistry integrated Modelling (ESCiMo) with the Modular Earth Submodel System (MESSy) version 2.51, Geosci. Model Dev., 9, 1153–1200, https://doi.org/10.5194/gmd-9-1153-2016, 2016. a, b
Johansson, L., Jalkanen, J.-P., Kalli, J., and Kukkonen, J.: The evolution of shipping emissions and the costs of regulation changes in the northern EU area, Atmos. Chem. Phys., 13, 11375–11389, https://doi.org/10.5194/acp-13-11375-2013, 2013. a
Johnson, M. S., Meskhidze, N., and Praju Kiliyanpilakkil, V.: A global comparison of GEOS-Chem-predicted and remotely-sensed mineral dust aerosol optical depth and extinction profiles, J. Adv. Model Earth Syst., 4, M07001, https://doi.org/10.1029/2011MS000109, 2012. a
Jonson, J. E., Jalkanen, J. P., Johansson, L., Gauss, M., and Denier van der Gon, H. A. C.: Model calculations of the effects of present and future emissions of air pollutants from shipping in the Baltic Sea and the North Sea, Atmos. Chem. Phys., 15, 783–798, https://doi.org/10.5194/acp-15-783-2015, 2015. a
Kaaden, N., Massling, A., Schladitz, A., Müller, T., Kandler, K., Schütz, L., Weinzierl, B., Petzold, A., Tesche, M., Leinert, S., Deutscher, C., Ebert, M., Weinbruch, S., and Wiedensohler, A.: State of mixing, shape factor, number size distribution, and hygroscopic growth of the Saharan anthropogenic and mineral dust aerosol at Tinfou, Morocco, Tellus B, 61, 51–63, https://doi.org/10.1111/j.1600-0889.2008.00388.x, 2009. a
Kaiser, C.: Including Coarse Mode Aerosol Microphysics in a Climate Model: Model Development and First Application, Ph.D. thesis, DLR-Forschungsbericht, DLR-FB-2016-1, 170 pp., Deutsches Zentrum für Luft- und Raumfahrt e.V., available at: http://elib.dlr.de/103328/ (last access: 9 January 2019), 2016. a, b
Kaiser, J. C., Hendricks, J., Righi, M., Riemer, N., Zaveri, R. A., Metzger, S., and Aquila, V.: The MESSy aerosol submodel MADE3 (v2.0b): description and a box model test, Geosci. Model Dev., 7, 1137–1157, https://doi.org/10.5194/gmd-7-1137-2014, 2014. a, b, c, d, e, f, g, h, i
Kandler, K., Lieke, K., Benker, N., Emmel, C., Küpper, M., Müller-Ebert, D., Ebert, M., Scheuvens, D., Schladitz, A., Schütz, L., and Weinbruch, S.: Electron microscopy of particles collected at Praia, Cape Verde, during the Saharan Mineral Dust Experiment: particle chemistry, shape, mixing state and complex refractive index, Tellus B, 63, 475–496, https://doi.org/10.1111/j.1600-0889.2011.00550.x, 2011. a, b
Kärcher, B. and Lohmann, U.: A Parameterization of cirrus cloud formation: Homogeneous freezing including effects of aerosol size, J. Geophys. Res.-Atmos., 107, AAC 9-1–AAC 9-10, https://doi.org/10.1029/2001JD001429, 2002. a
Kerkweg, A., Buchholz, J., Ganzeveld, L., Pozzer, A., Tost, H., and Jöckel, P.: Technical Note: An implementation of the dry removal processes DRY DEPosition and SEDImentation in the Modular Earth Submodel System (MESSy), Atmos. Chem. Phys., 6, 4617–4632, https://doi.org/10.5194/acp-6-4617-2006, 2006a. a, b
Kerkweg, A., Sander, R., Tost, H., and Jöckel, P.: Technical note: Implementation of prescribed (OFFLEM), calculated (ONLEM), and pseudo-emissions (TNUDGE) of chemical species in the Modular Earth Submodel System (MESSy), Atmos. Chem. Phys., 6, 3603–3609, https://doi.org/10.5194/acp-6-3603-2006, 2006b. a, b, c
Kerkweg, A., Buchholz, J., Ganzeveld, L., Pozzer, A., Tost, H., and Jöckel, P.: Corrigendum to ”Technical Note: An implementation of the dry removal processes DRY DEPosition and SEDImentation in the Modular Earth Submodel System (MESSy)” published in Atmos. Chem. Phys., 6, 4617–4632, 2006, Atmos. Chem. Phys., 9, 9569–9569, https://doi.org/10.5194/acp-9-9569-2009, 2009. a
Kerminen, V. M., Pakkanen, T. A., and Hillamo, R. E.: Interactions between inorganic trace gases and supermicrometer particles at a coastal site, Atmos. Environ., 31, 2753–2765, https://doi.org/10.1016/S1352-2310(97)00092-7, 1997. a
Kerminen, V.-M., Petäjä, T., Manninen, H. E., Paasonen, P., Nieminen, T., Sipilä, M., Junninen, H., Ehn, M., Gagné, S., Laakso, L., Riipinen, I., Vehkamäki, H., Kurten, T., Ortega, I. K., Dal Maso, M., Brus, D., Hyvärinen, A., Lihavainen, H., Leppä, J., Lehtinen, K. E. J., Mirme, A., Mirme, S., Hõrrak, U., Berndt, T., Stratmann, F., Birmili, W., Wiedensohler, A., Metzger, A., Dommen, J., Baltensperger, U., Kiendler-Scharr, A., Mentel, T. F., Wildt, J., Winkler, P. M., Wagner, P. E., Petzold, A., Minikin, A., Plass-Dülmer, C., Pöschl, U., Laaksonen, A., and Kulmala, M.: Atmospheric nucleation: highlights of the EUCAARI project and future directions, Atmos. Chem. Phys., 10, 10829–10848, https://doi.org/10.5194/acp-10-10829-2010, 2010. a
Khalizov, A. F., Zhang, R., Zhang, D., Xue, H., Pagels, J., and McMurry, P. H.: Formation of highly hygroscopic soot aerosols upon internal mixing with sulfuric acid vapor, J. Geophys. Res.-Atmos., 114, D05208, https://doi.org/10.1029/2008JD010595, 2009. a
Kipling, Z., Stier, P., Schwarz, J. P., Perring, A. E., Spackman, J. R., Mann, G. W., Johnson, C. E., and Telford, P. J.: Constraints on aerosol processes in climate models from vertically-resolved aircraft observations of black carbon, Atmos. Chem. Phys., 13, 5969–5986, https://doi.org/10.5194/acp-13-5969-2013, 2013. a
Kirkby, J., Duplissy, J., Sengupta, K., Frege, C., Gordon, H., Williamson, C., Heinritzi, M., Simon, M., Yan, C., Almeida, J., Tröstl, J., Nieminen, T., Ortega, I. K., Wagner, R., Adamov, A., Amorim, A., Bernhammer, A.-K., Bianchi, F., Breitenlechner, M., Brilke, S., Chen, X., Craven, J., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Hakala, J., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Mathot, S., Molteni, U., Onnela, A., Peräkylä, O., Piel, F., Petäjä, T., Praplan, A. P., Pringle, K., Rap, A., Richards, N. A. D., Riipinen, I., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Scott, C. E., Seinfeld, J. H., Sipilä, M., Steiner, G., Stozhkov, Y., Stratmann, F., Tomè, A., Virtanen, A., Vogel, A. L., Wagner, A. C., Wagner, P. E., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Zhang, X., Hansel, A., Dommen, J., Donahue, N. M., Worsnop, D. R., Baltensperger, U., Kulmala, M., Carslaw, K. S., and Curtius, J.: Ion-induced nucleation of pure biogenic particles, Nature, 533, 521–526, https://doi.org/10.1038/nature17953, 2016. a
Kirkevåg, A., Iversen, T., Seland, Ø., Hoose, C., Kristjánsson, J. E., Struthers, H., Ekman, A. M. L., Ghan, S., Griesfeller, J., Nilsson, E. D., and Schulz, M.: Aerosol–climate interactions in the Norwegian Earth System Model – NorESM1-M, Geosci. Model Dev., 6, 207–244, https://doi.org/10.5194/gmd-6-207-2013, 2013. a, b, c
Koch, D., Schulz, M., Kinne, S., McNaughton, C., Spackman, J. R., Balkanski, Y., Bauer, S., Berntsen, T., Bond, T. C., Boucher, O., Chin, M., Clarke, A., De Luca, N., Dentener, F., Diehl, T., Dubovik, O., Easter, R., Fahey, D. W., Feichter, J., Fillmore, D., Freitag, S., Ghan, S., Ginoux, P., Gong, S., Horowitz, L., Iversen, T., Kirkevåg, A., Klimont, Z., Kondo, Y., Krol, M., Liu, X., Miller, R., Montanaro, V., Moteki, N., Myhre, G., Penner, J. E., Perlwitz, J., Pitari, G., Reddy, S., Sahu, L., Sakamoto, H., Schuster, G., Schwarz, J. P., Seland, Ø., Stier, P., Takegawa, N., Takemura, T., Textor, C., van Aardenne, J. A., and Zhao, Y.: Evaluation of black carbon estimations in global aerosol models, Atmos. Chem. Phys., 9, 9001–9026, https://doi.org/10.5194/acp-9-9001-2009, 2009. a
Koch, D., Schulz, M., Kinne, S., McNaughton, C., Spackman, J. R., Balkanski, Y., Bauer, S., Berntsen, T., Bond, T. C., Boucher, O., Chin, M., Clarke, A., De Luca, N., Dentener, F., Diehl, T., Dubovik, O., Easter, R., Fahey, D. W., Feichter, J., Fillmore, D., Freitag, S., Ghan, S., Ginoux, P., Gong, S., Horowitz, L., Iversen, T., Kirkevåg, A., Klimont, Z., Kondo, Y., Krol, M., Liu, X., Miller, R., Montanaro, V., Moteki, N., Myhre, G., Penner, J. E., Perlwitz, J., Pitari, G., Reddy, S., Sahu, L., Sakamoto, H., Schuster, G., Schwarz, J. P., Seland, Ø., Stier, P., Takegawa, N., Takemura, T., Textor, C., van Aardenne, J. A., and Zhao, Y.: Corrigendum to ”Evaluation of black carbon estimations in global aerosol models” published in Atmos. Chem. Phys., 9, 9001–9026, 2009, Atmos. Chem. Phys., 10, 79–81, https://doi.org/10.5194/acp-10-79-2010, 2010. a
Korolev, A. V., Isaac, G. A., Cober, S. G., Strapp, J. W., and Hallett, J.: Microphysical characterization of mixed-phase clouds, Q. J. Roy. Meteorol. Soc., 129, 39–65, https://doi.org/10.1256/qj.01.204, 2003. a
Kuebbeler, M., Lohmann, U., Hendricks, J., and Kärcher, B.: Dust ice nuclei effects on cirrus clouds, Atmos. Chem. Phys., 14, 3027–3046, https://doi.org/10.5194/acp-14-3027-2014, 2014. a
Lamarque, J.-F., Bond, T. C., Eyring, V., Granier, C., Heil, A., Klimont, Z., Lee, D. S., Liousse, C., Mieville, A., Owen, B., Schultz, M. G., Shindell, D. T., Smith, S. J., Stehfest, E., van Aardenne, J. A., Cooper, O. R., Kainuma, M., Mahowald, N., McConnell, J. R., Naik, V., Riahi, K., and van Vuuren, D. P.: Historical (1850–2000) gridded anthropogenic and biomass burning emissions of reactive gases and aerosols: methodology and application, Atmos. Chem. Phys., 10, 7017–7039, https://doi.org/10.5194/acp-10-7017-2010, 2010. a, b
Lana, A., Bell, T. G., Simo, R., Vallina, S. M., Ballabrera-Poy, J., Kettle, A. J., Dachs, J., Bopp, L., Saltzman, E. S., Stefels, J., Johnson, J. E., and Liss, P. S.: An updated climatology of surface dimethlysulfide concentrations and emission fluxes in the global ocean, Global Biogeochem. Cy., 25, GB1004, https://doi.org/10.1029/2010GB003850, 2011. a
Lauer, A.: Untersuchung von Größenverteilung und Zusammensetzung des troposphärischen Aerosols mit einem globalen Zirkulationsmodell, Ph.D. thesis, FU Berlin, Germany, 2004. a
Lauer, A. and Hendricks, J.: Simulating aerosol microphysics with the ECHAM4/MADE GCM – Part II: Results from a first multiannual simulation of the submicrometer aerosol, Atmos. Chem. Phys., 6, 5495–5513, https://doi.org/10.5194/acp-6-5495-2006, 2006. a, b
Lauer, A., Eyring, V., Hendricks, J., Jöckel, P., and Lohmann, U.: Global model simulations of the impact of ocean-going ships on aerosols, clouds, and the radiation budget, Atmos. Chem. Phys., 7, 5061–5079, https://doi.org/10.5194/acp-7-5061-2007, 2007. a, b, c, d
Lauer, A., Eyring, V., Corbett, J. J., Wang, C., and Winebrake, J. J.: Assessment of Near-Future Policy Instruments for Oceangoing Shipping: Impact on Atmospheric Aerosol Burdens and the Earth's Radiation Budget, Environ. Sci. Tech., 43, 5592–5598, https://doi.org/10.1021/es900922h, 2009. a, b, c, d
Lauer, A., Eyring, V., Righi, M., Buchwitz, M., Defourny, P., Evaldsson, M., Friedlingstein, P., de Jeu, R., de Leeuw, G., Loew, A., Merchant, C. J., Müller, B., Popp, T., Reuter, M., Sandven, S., Senftleben, D., Stengel, M., Roozendael, M. V., Wenzel, S., and Willén, U.: Benchmarking CMIP5 models with a subset of ESA CCI Phase 2 data using the ESMValTool, Remote Sens. Environ., 203, 9–39, https://doi.org/10.1016/j.rse.2017.01.007, 2017. a
Lee, L. A., Pringle, K. J., Reddington, C. L., Mann, G. W., Stier, P., Spracklen, D. V., Pierce, J. R., and Carslaw, K. S.: The magnitude and causes of uncertainty in global model simulations of cloud condensation nuclei, Atmos. Chem. Phys., 13, 8879–8914, https://doi.org/10.5194/acp-13-8879-2013, 2013a. a
Lee, L. A., Pringle, K. J., Reddington, C. L., Mann, G. W., Stier, P., Spracklen, D. V., Pierce, J. R., and Carslaw, K. S.: Corrigendum to ”The magnitude and causes of uncertainty in global model simulations of cloud condensation nuclei” published in Atmos. Chem. Phys., 13, 8879–8914, 2013, Atmos. Chem. Phys., 13, 9375–9377, https://doi.org/10.5194/acp-13-9375-2013, 2013b. a
Lee, Y. H., Adams, P. J., and Shindell, D. T.: Evaluation of the global aerosol microphysical ModelE2-TOMAS model against satellite and ground-based observations, Geosci. Model Dev., 8, 631–667, https://doi.org/10.5194/gmd-8-631-2015, 2015. a, b, c, d, e
Levy, R. C., Mattoo, S., Munchak, L. A., Remer, L. A., Sayer, A. M., Patadia, F., and Hsu, N. C.: The Collection 6 MODIS aerosol products over land and ocean, Atmos. Meas. Tech., 6, 2989–3034, https://doi.org/10.5194/amt-6-2989-2013, 2013. a
Li, J., Han, Z., and Zhang, R.: Influence of aerosol hygroscopic growth parameterization on aerosol optical depth and direct radiative forcing over East Asia, Atmos. Res., 140–141, 14–27, https://doi.org/10.1016/j.atmosres.2014.01.013, 2014. a
Liss, P. S. and Merlivat, L.: Air-Sea Gas Exchange Rates: Introduction and Synthesis, in: The Role of Air-Sea Exchange in Geochemical Cycling, edited by Buat-Ménard, P., Vol. 185 of NATO ASI Series, 113–127, Springer Netherlands, https://doi.org/10.1007/978-94-009-4738-2_5, 1986. a
Liu, D., Allan, J., Whitehead, J., Young, D., Flynn, M., Coe, H., McFiggans, G., Fleming, Z. L., and Bandy, B.: Ambient black carbon particle hygroscopic properties controlled by mixing state and composition, Atmos. Chem. Phys., 13, 2015–2029, https://doi.org/10.5194/acp-13-2015-2013, 2013. a
Lohmann, U. and Feichter, J.: Global indirect aerosol effects: a review, Atmos. Chem. Phys., 5, 715–737, https://doi.org/10.5194/acp-5-715-2005, 2005. a
Lohmann, U. and Hoose, C.: Sensitivity studies of different aerosol indirect effects in mixed-phase clouds, Atmos. Chem. Phys., 9, 8917–8934, https://doi.org/10.5194/acp-9-8917-2009, 2009. a, b
Lohmann, U., Feichter, J., Chuang, C. C., and Penner, J. E.: Prediction of the number of cloud droplets in the ECHAM GCM, J. Geophys. Res.-Atmos., 104, 9169–9198, https://doi.org/10.1029/1999JD900046, 1999. a
Lohmann, U., Kärcher, B., and Hendricks, J.: Sensitivity studies of cirrus clouds formed by heterogeneous freezing in the ECHAM GCM, J. Geophys. Res. Atmos., 109, d16204, https://doi.org/10.1029/2003JD004443, 2004. a
Lohmann, U., Stier, P., Hoose, C., Ferrachat, S., Kloster, S., Roeckner, E., and Zhang, J.: Cloud microphysics and aerosol indirect effects in the global climate model ECHAM5-HAM, Atmos. Chem. Phys., 7, 3425–3446, https://doi.org/10.5194/acp-7-3425-2007, 2007. a
Lund, M. T., Eyring, V., Fuglestvedt, J., Hendricks, J., Lauer, A., Lee, D., and Righi, M.: Global-Mean Temperature Change from Shipping toward 2050: Improved Representation of the Indirect Aerosol Effect in Simple Climate Models, Environ. Sci. Tech., 46, 8868–8877, https://doi.org/10.1021/es301166e, 2012. a
Lund, M. T., Berntsen, T. K., and Samset, B. H.: Sensitivity of black carbon concentrations and climate impact to aging and scavenging in OsloCTM2–M7, Atmos. Chem. Phys., 17, 6003–6022, https://doi.org/10.5194/acp-17-6003-2017, 2017. a
Mahowald, N., Albani, S., Kok, J. F., Engelstaeder, S., Scanza, R., Ward, D. S., and Flanner, M. G.: The size distribution of desert dust aerosols and its impact on the Earth system, Aeolian Res., 15, 53–71, https://doi.org/10.1016/j.aeolia.2013.09.002, 2014. a
Makkonen, R., Seland, Ø., Kirkevåg, A., Iversen, T., and Kristjánsson, J. E.: Evaluation of aerosol number concentrations in NorESM with improved nucleation parameterization, Atmos. Chem. Phys., 14, 5127–5152, https://doi.org/10.5194/acp-14-5127-2014, 2014. a
Mann, G. W., Carslaw, K. S., Spracklen, D. V., Ridley, D. A., Manktelow, P. T., Chipperfield, M. P., Pickering, S. J., and Johnson, C. E.: Description and evaluation of GLOMAP-mode: a modal global aerosol microphysics model for the UKCA composition-climate model, Geosci. Model Dev., 3, 519–551, https://doi.org/10.5194/gmd-3-519-2010, 2010. a, b
Massling, A., Nielsen, I. E., Kristensen, D., Christensen, J. H., Sørensen, L. L., Jensen, B., Nguyen, Q. T., Nøjgaard, J. K., Glasius, M., and Skov, H.: Atmospheric black carbon and sulfate concentrations in Northeast Greenland, Atmos. Chem. Phys., 15, 9681–9692, https://doi.org/10.5194/acp-15-9681-2015, 2015. a
Matsui, H., Koike, M., Kondo, Y., Takegawa, N., Wiedensohler, A., Fast, J. D., and Zaveri, R. A.: Impact of new particle formation on the concentrations of aerosols and cloud condensation nuclei around Beijing, J. Geophys. Res.-Atmos., 116, D19208, https://doi.org/10.1029/2011JD016025, 2011. a
Matsui, H., Koike, M., Kondo, Y., Moteki, N., Fast, J. D., and Zaveri, R. A.: Development and validation of a black carbon mixing state resolved three-dimensional model: Aging processes and radiative impact, J. Geophys. Res.-Atmos., 118, 2304–2326, https://doi.org/10.1029/2012JD018446, 2013. a
Mayer, B. and Kylling, A.: Technical note: The libRadtran software package for radiative transfer calculations – description and examples of use, Atmos. Chem. Phys., 5, 1855–1877, https://doi.org/10.5194/acp-5-1855-2005, 2005. a
Michou, M., Nabat, P., and Saint-Martin, D.: Development and basic evaluation of a prognostic aerosol scheme (v1) in the CNRM Climate Model CNRM-CM6, Geosci. Model Dev., 8, 501–531, https://doi.org/10.5194/gmd-8-501-2015, 2015. a, b
Minikin, A., Petzold, A., Strom, J., Krejci, R., Seifert, M., van Velthoven, P., Schlager, H., and Schumann, U.: Aircraft observations of the upper tropospheric fine particle aerosol in the Northern and Southern Hemispheres at midlatitudes, Geophys. Res. Lett., 30, 1503, https://doi.org/10.1029/2002GL016458, 2003. a
Modini, R. L., Ristovski, Z. D., Johnson, G. R., He, C., Surawski, N., Morawska, L., Suni, T., and Kulmala, M.: New particle formation and growth at a remote, sub-tropical coastal location, Atmos. Chem. Phys., 9, 7607–7621, https://doi.org/10.5194/acp-9-7607-2009, 2009. a
Moore, R. H., Thornhill, K. L., Weinzierl, B., Sauer, D., D'Ascoli, E., Kim, J., Lichtenstern, M., Scheibe, M., Beaton, B., Beyersdorf, A. J., Barrick, J., Bulzan, D., Corr, C. A., Crosbie, E., Jurkat, T., Martin, R., Riddick, D., Shook, M., Slover, G., Voigt, C., White, R., Winstead, E., Yasky, R., Ziemba, L. D., Brown, A., Schlager, H., and Anderson, B. E.: Biofuel blending reduces particle emissions from aircraft engines at cruise conditions, Nature, 543, 411–415, https://doi.org/10.1038/nature21420, 2017. a
Nabat, P., Solmon, F., Mallet, M., Kok, J. F., and Somot, S.: Dust emission size distribution impact on aerosol budget and radiative forcing over the Mediterranean region: a regional climate model approach, Atmos. Chem. Phys., 12, 10545–10567, https://doi.org/10.5194/acp-12-10545-2012, 2012. a
Network Center for EANET: Data Report on the Acid Deposition in the East Asian Region 2013, Tech. rep., Asia Center for Air Pollution Research (ACAP), Niigata-shi, Japan, available at: http://www.eanet.asia/product/datarep/datarep13/datarep13.pdf (last access: 9 January 2019), 2014. a
Nolte, C. G., Bhave, P. V., Arnold, J. R., Dennis, R. L., Zhang, K. M., and Wexler, A. S.: Modeling urban and regional aerosols – Application of the CMAQ-UCD Aerosol Model to Tampa, a coastal urban site, Atmos. Environ., 42, 3179–3191, https://doi.org/10.1016/j.atmosenv.2007.12.059, 2008. a
Nordeng, T. E.: Extended versions of the convective parameterization scheme at ECMWF and their impact on the mean and transient activity of the model in the tropics, Technical Memorandum 206, European Centre for Medium-Range Weather Forecasts, Reading, UK, https://doi.org/10.21957/e34xwhysw, 1994. a
North, P., Briggs, S. A., Plummer, S. E., and Settle, J. J.: Retrieval of land surface bidirectional reflectance and aerosol opacity from ATSR-2 multiangle imagery, IEEE Trans.Geosci. Rem. Sens., 37, 526–537, https://doi.org/10.1109/36.739106, 1999. a
Olivié, D. J. L., Cariolle, D., Teyssèdre, H., Salas, D., Voldoire, A., Clark, H., Saint-Martin, D., Michou, M., Karcher, F., Balkanski, Y., Gauss, M., Dessens, O., Koffi, B., and Sausen, R.: Modeling the climate impact of road transport, maritime shipping and aviation over the period 1860–2100 with an AOGCM, Atmos. Chem. Phys., 12, 1449–1480, https://doi.org/10.5194/acp-12-1449-2012, 2012. a
Ouwersloot, H. G., Pozzer, A., Steil, B., Tost, H., and Lelieveld, J.: Revision of the convective transport module CVTRANS 2.4 in the EMAC atmospheric chemistry–climate model, Geosci. Model Dev., 8, 2435–2445, https://doi.org/10.5194/gmd-8-2435-2015, 2015. a
Peters, K., Stier, P., Quaas, J., and Graßl, H.: Aerosol indirect effects from shipping emissions: sensitivity studies with the global aerosol-climate model ECHAM-HAM, Atmos. Chem. Phys., 12, 5985–6007, https://doi.org/10.5194/acp-12-5985-2012, 2012. a
Peters, K., Stier, P., Quaas, J., and Graßl, H.: Corrigendum to ”Aerosol indirect effects from shipping emissions: sensitivity studies with the global aerosol-climate model ECHAM-HAM” published in Atmos. Chem. Phys., 12, 5985–6007, 2012, Atmos. Chem. Phys., 13, 6429–6430, https://doi.org/10.5194/acp-13-6429-2013, 2013. a
Petzold, A., Döpelheuer, A., Brock, C. A., and Schröder, F.: In situ observations and model calculations of black carbon emission by aircraft at cruise altitude, J. Geophys. Res.-Atmos., 104, 22171–22181, https://doi.org/10.1029/1999JD900460, 1999. a
Petzold, A., Fiebig, M., Flentje, H., Keil, A., Leiterer, U., Schröder, F., Stifter, A., Wendisch, M., and Wendling, P.: Vertical variability of aerosol properties observed at a continental site during the Lindenberg Aerosol Characterization Experiment (LACE 98), J. Geophys. Res.-Atmos., 107, 8128, https://doi.org/10.1029/2001JD001043, 2002. a, b
Petzold, A., Hasselbach, J., Lauer, P., Baumann, R., Franke, K., Weingartner, E., Gurk, C., and Schlager, H.: Experimental studies on particle emissions from cruising ship, their characteristic properties, transformation and atmospheric lifetime in the marine boundary layer, Atmos. Chem. Phys., 8, 2387–2403, https://doi.org/10.5194/acp-8-2387-2008, 2008. a
Petzold, A., Ogren, J. A., Fiebig, M., Laj, P., Li, S.-M., Baltensperger, U., Holzer-Popp, T., Kinne, S., Pappalardo, G., Sugimoto, N., Wehrli, C., Wiedensohler, A., and Zhang, X.-Y.: Recommendations for reporting ”black carbon” measurements, Atmos. Chem. Phys., 13, 8365–8379, https://doi.org/10.5194/acp-13-8365-2013, 2013. a, b, c
Pierce, J. R., Croft, B., Kodros, J. K., D'Andrea, S. D., and Martin, R. V.: The importance of interstitial particle scavenging by cloud droplets in shaping the remote aerosol size distribution and global aerosol-climate effects, Atmos. Chem. Phys., 15, 6147–6158, https://doi.org/10.5194/acp-15-6147-2015, 2015. a
Pietikäinen, J.-P., Mikkonen, S., Hamed, A., Hienola, A. I., Birmili, W., Kulmala, M., and Laaksonen, A.: Analysis of nucleation events in the European boundary layer using the regional aerosol–climate model REMO-HAM with a solar radiation-driven OH-proxy, Atmos. Chem. Phys., 14, 11711–11729, https://doi.org/10.5194/acp-14-11711-2014, 2014. a
Popp, T., de Leeuw, G., Bingen, C., Brühl, C., Capelle, V., Chedin, A., Clarisse, L., Dubovik, O., Grainger, R., Griesfeller, J., Heckel, A., Kinne, S., Klüser, L., Kosmale, M., Kolmonen, P., Lelli, L., Litvinov, P., Mei, L., North, P., Pinnock, S., Povey, A., Robert, C., Schulz, M., Sogacheva, L., Stebel, K., Stein Zweers, D., Thomas, G., Tilstra, L. G., Vandenbussche, S., Veefkind, P., Vountas, M., and Xue, Y.: Development, Production and Evaluation of Aerosol Climate Data Records from European Satellite Observations (Aerosol_cci), Remote Sens., 8, https://doi.org/10.3390/rs8050421, 2016. a
Pozzer, A., de Meij, A., Pringle, K. J., Tost, H., Doering, U. M., van Aardenne, J., and Lelieveld, J.: Distributions and regional budgets of aerosols and their precursors simulated with the EMAC chemistry-climate model, Atmos. Chem. Phys., 12, 961–987, https://doi.org/10.5194/acp-12-961-2012, 2012. a, b, c, d, e, f
Pozzer, A., de Meij, A., Yoon, J., Tost, H., Georgoulias, A. K., and Astitha, M.: AOD trends during 2001–2010 from observations and model simulations, Atmos. Chem. Phys., 15, 5521–5535, https://doi.org/10.5194/acp-15-5521-2015, 2015. a
Prabhakar, G., Ervens, B., Wang, Z., Maudlin, L. C., Coggon, M. M., Jonsson, H. H., Seinfeld, J. H., and Sorooshian, A.: Sources of nitrate in stratocumulus cloud water: Airborne measurements during the 2011 E-PEACE and 2013 NiCE studies, Atmos. Environ., 97, 166–173, https://doi.org/10.1016/j.atmosenv.2014.08.019, 2014. a
Price, C. and Rind, D.: A simple lightning parameterization for calculating global lightning distributions, J. Geophys. Res.-Atmos., 97, 9919–9933, https://doi.org/10.1029/92JD00719, 1992. a
Pringle, K. J., Tost, H., Message, S., Steil, B., Giannadaki, D., Nenes, A., Fountoukis, C., Stier, P., Vignati, E., and Lelieveld, J.: Description and evaluation of GMXe: a new aerosol submodel for global simulations (v1), Geosci. Model Dev., 3, 391–412, https://doi.org/10.5194/gmd-3-391-2010, 2010. a
Pruppacher, H. R. and Klett, J. D.: Microphysics of Clouds and Precipitation, Atmospheric and Oceanographic Sciences Library, Kluwer Academic Publishers, Dordrecht, The Netherlands, second edn., 1997. a
Putaud, J.-P., Van Dingenen, R., Baltensperger, U., Brüggemann, E., Charron, A., Facchini, M., Decesari, S., Fuzzi, S., Gehrig, R., Hansson, H.-C., Harrison, R., Jones, A., Laj, P., Lorbeer, G., Maenhaut, W., Mihalopoulos, N., Müller, K., Palmgren, F., Querol, X., Rodriguez, S., Schneider, J., Spindler, G., ten Brink, H., Tunved, P., Tørseth, K., Wehner, B., Weingartner, E., Wiedensohler, A., Wåhlin, P., and Raes, F.: A European Aerosol Phenomenology, Tech. Rep. EUR 20411 EN, European Commission, Joint Research Centre, Institute for Environment and Sustainability, Ispra, Italy, 2003. a, b, c, d
Righi, M.: Evaluation of the global aerosol model EMAC-MADE3 in the uncoupled version – monthly data, World Data Center for Climate (WDCC) at DKRZ, https://doi.org/10.1594/WDCC/EMAC-MADE3_monthly, 2018a. a
Righi, M.: Evaluation of the global aerosol model EMAC-MADE3 in the uncoupled version – 12 h data, World Data Center for Climate (WDCC) at DKRZ, https://doi.org/10.1594/WDCC/EMAC-MADE3_12h, 2018b. a
Righi, M., Klinger, C., Eyring, V., Hendricks, J., Lauer, A., and Petzold, A.: Climate Impact of Biofuels in Shipping: Global Model Studies of the Aerosol Indirect Effect, Environ. Sci. Technol., 45, 3519–3525, https://doi.org/10.1021/es1036157, 2011. a, b, c, d
Righi, M., Hendricks, J., and Sausen, R.: The global impact of the transport sectors on atmospheric aerosol: simulations for year 2000 emissions, Atmos. Chem. Phys., 13, 9939–9970, https://doi.org/10.5194/acp-13-9939-2013, 2013. a, b, c, d, e, f, g
Righi, M., Hendricks, J., and Sausen, R.: The global impact of the transport sectors on atmospheric aerosol in 2030 – Part 1: Land transport and shipping, Atmos. Chem. Phys., 15, 633–651, https://doi.org/10.5194/acp-15-633-2015, 2015. a
Righi, M., Hendricks, J., and Sausen, R.: The global impact of the transport sectors on atmospheric aerosol in 2030 – Part 2: Aviation, Atmos. Chem. Phys., 16, 4481–4495, https://doi.org/10.5194/acp-16-4481-2016, 2016. a
Roeckner, E., Bäuml, G., Bonaventura, L., Brokopf, R., Esch, M., Giorgetta, M., Hagemann, S., Kirchner, I., Kornblueh, L., Manzini, E., Rhodin, A., Schlese, U., Schulzweida, U., and Tompkins, A.: The atmospheric general circulation model ECHAM 5. PART I: Model description, Tech. Rep. 349, Max-Planck-Institut für Meteorologie, Hamburg, Germany, available at: http://www.mpimet.mpg.de/fileadmin/publikationen/Reports/max_scirep_349.pdf (last access: 9 January 2019), 2003. a, b, c
Roeckner, E., Brokopf, R., Esch, M., Giorgetta, M., Hagemann, S., Kornblueh, L., Manzini, E., Schlese, U., and Schulzweida, U.: Sensitivity of Simulated Climate to Horizontal and Vertical Resolution in the ECHAM5 Atmosphere Model, J. Climate, 19, 3771–3791, https://doi.org/10.1175/JCLI3824.1, 2006. a, b
Roiger, A., Thomas, J.-L., Schlager, H., Law, K. S., Kim, J., Schäfler, A., Weinzierl, B., Dahlkötter, F., Krisch, I., Marelle, L., Minikin, A., Raut, J.-C., Reiter, A., Rose, M., Scheibe, M., Stock, P., Baumann, R., Bouarar, I., Clerbaux, C., George, M., Onishi, T., and Flemming, J.: Quantifying Emerging Local Anthropogenic Emissions in the Arctic Region: The ACCESS Aircraft Campaign Experiment, Bull. Am. Meteorol. Soc., 96, 441–460, https://doi.org/10.1175/BAMS-D-13-00169.1, 2015. a
Sander, R., Baumgaertner, A., Gromov, S., Harder, H., Jöckel, P., Kerkweg, A., Kubistin, D., Regelin, E., Riede, H., Sandu, A., Taraborrelli, D., Tost, H., and Xie, Z.-Q.: The atmospheric chemistry box model CAABA/MECCA-3.0, Geosci. Model Dev., 4, 373–380, https://doi.org/10.5194/gmd-4-373-2011, 2011. a
Sander, R., Jöckel, P., Kirner, O., Kunert, A. T., Landgraf, J., and Pozzer, A.: The photolysis module JVAL-14, compatible with the MESSy standard, and the JVal PreProcessor (JVPP), Geosci. Model Dev., 7, 2653–2662, https://doi.org/10.5194/gmd-7-2653-2014, 2014. a
Schembari, C., Cavalli, F., Cuccia, E., Hjorth, J., Calzolai, G., Pérez, N., Pey, J., Prati, P., and Raes, F.: Impact of a European directive on ship emissions on air quality in Mediterranean harbours, Atmos. Environ., 61, 661–669, https://doi.org/10.1016/j.atmosenv.2012.06.047, 2012. a
Schumann, U. and Huntrieser, H.: The global lightning-induced nitrogen oxides source, Atmos. Chem. Phys., 7, 3823–3907, https://doi.org/10.5194/acp-7-3823-2007, 2007. a
Schutgens, N. A. J., Gryspeerdt, E., Weigum, N., Tsyro, S., Goto, D., Schulz, M., and Stier, P.: Will a perfect model agree with perfect observations? The impact of spatial sampling, Atmos. Chem. Phys., 16, 6335–6353, https://doi.org/10.5194/acp-16-6335-2016, 2016a. a
Schutgens, N. A. J., Partridge, D. G., and Stier, P.: The importance of temporal collocation for the evaluation of aerosol models with observations, Atmos. Chem. Phys., 16, 1065–1079, https://doi.org/10.5194/acp-16-1065-2016, 2016b. a, b, c
Schwarz, J. P., Gao, R. S., Fahey, D. W., Thomson, D. S., Watts, L. A., Wilson, J. C., Reeves, J. M., Darbeheshti, M., Baumgardner, D. G., Kok, G. L., Chung, S. H., Schulz, M., Hendricks, J., Lauer, A., Kärcher, B., Slowik, J. G., Rosenlof, K. H., Thompson, T. L., Langford, A. O., Loewenstein, M., and Aikin, K. C.: Single-particle measurements of midlatitude black carbon and light-scattering aerosols from the boundary layer to the lower stratosphere, J. Geophys. Res.-Atmos., 111, D16207, https://doi.org/10.1029/2006JD007076, 2006. a, b
Schwarz, J. P., Spackman, J. R., Fahey, D. W., Gao, R. S., Lohmann, U., Stier, P., Watts, L. A., Thomson, D. S., Lack, D. A., Pfister, L., Mahoney, M. J., Baumgardner, D., Wilson, J. C., and Reeves, J. M.: Coatings and their enhancement of black carbon light absorption in the tropical atmosphere, J. Geophys. Res.-Atmos., 113, D03203, https://doi.org/10.1029/2007JD009042, 2008. a
Schwarz, J. P., Samset, B. H., Perring, A. E., Spackman, J. R., Gao, R. S., Stier, P., Schulz, M., Moore, F. L., Ray, E. A., and Fahey, D. W.: Global-scale seasonally resolved black carbon vertical profiles over the Pacific, Geophys. Res. Lett., 40, 5542–5547, https://doi.org/10.1002/2013GL057775, 2013. a, b, c
Schwarz, J. P., Weinzierl, B., Samset, B. H., Dollner, M., Heimerl, K., Markovic, M. Z., Perring, A. E., and Ziemba, L.: Aircraft measurements of black carbon vertical profiles show upper tropospheric variability and stability, Geophys. Res. Lett., 44, 1132–1140, https://doi.org/10.1002/2016GL071241, 2016GL071241, 2017. a, b, c, d, e
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics, John Wiley and Sons, New York, US, 1998. a
Sellers, P., Randall, D., Collatz, G., Berry, J., Field, C., Dazlich, D., Zhang, C., Collelo, G., and Bounoua, L.: A Revised Land Surface Parameterization (SiB2) for Atmospheric GCMS. Part I: Model Formulation, J. Climate, 9, 676–705, https://doi.org/10.1175/1520-0442(1996)009<0676:ARLSPF>2.0.CO;2, 1996. a
Slinn, W. G. N.: Precipitation Scavenging, in: Atmospheric science and power production, edited by: Randerson, D., chap. 11, 466–532, Department of Energy, U.S., https://doi.org/10.2172/6503687, 1984. a
Spackman, J. R., Gao, R. S., Schwarz, J. P., Watts, L. A., Fahey, D. W., Pfister, L., and Bui, T. P.: Seasonal variability of black carbon mass in the tropical tropopause layer, Geophys. Res. Lett., 38, L09803, https://doi.org/10.1029/2010GL046343, 2011. a
Spiro, P. A., Jacob, D. J., and Logan, J. A.: Global inventory of sulfur emissions with 1∘ × 1∘ resolution, J. Geophys. Res., 97, 6023–6036, https://doi.org/10.1029/91JD03139, 1992. a
Spracklen, D. V., Carslaw, K. S., Kulmala, M., Kerminen, V.-M., Mann, G. W., and Sihto, S.-L.: The contribution of boundary layer nucleation events to total particle concentrations on regional and global scales, Atmos. Chem. Phys., 6, 5631–5648, https://doi.org/10.5194/acp-6-5631-2006, 2006. a
Sundqvist, H., Berge, E., and Kristjánsson, J. E.: Condensation and Cloud Parameterization Studies with a Mesoscale Numerical Weather Prediction Model, Mon. Weather Rev., 117, 1641–1657, https://doi.org/10.1175/1520-0493(1989)117<1641:CACPSW>2.0.CO;2, 1989. a
Svenningsson, B., Hansson, H.-C., Wiedensohler, A., Noone, K., Ogren, J., Hallberg, A., and Colvile, R.: Hygroscopic growth of aerosol particles and its influence on nucleation scavenging in cloud: Experimental results from Kleiner Feldberg, J. Atmos. Chem., 19, 129–152, https://doi.org/10.1007/BF00696586, 1994. a
Textor, C., Schulz, M., Guibert, S., Kinne, S., Balkanski, Y., Bauer, S., Berntsen, T., Berglen, T., Boucher, O., Chin, M., Dentener, F., Diehl, T., Easter, R., Feichter, H., Fillmore, D., Ghan, S., Ginoux, P., Gong, S., Grini, A., Hendricks, J., Horowitz, L., Huang, P., Isaksen, I., Iversen, I., Kloster, S., Koch, D., Kirkevåg, A., Kristjansson, J. E., Krol, M., Lauer, A., Lamarque, J. F., Liu, X., Montanaro, V., Myhre, G., Penner, J., Pitari, G., Reddy, S., Seland, Ø., Stier, P., Takemura, T., and Tie, X.: Analysis and quantification of the diversities of aerosol life cycles within AeroCom, Atmos. Chem. Phys., 6, 1777–1813, https://doi.org/10.5194/acp-6-1777-2006, 2006. a
Tiedtke, M.: A Comprehensive Mass Flux Scheme for Cumulus Parameterization in Large-Scale Models, Mon. Weather Rev., 117, 1779–1800, https://doi.org/10.1175/1520-0493(1989)117<1779:ACMFSF>2.0.CO;2, 1989. a
Tompkins, A. M.: A Prognostic Parameterization for the Subgrid-Scale Variability of Water Vapor and Clouds in Large-Scale Models and Its Use to Diagnose Cloud Cover, J. Atmos. Sci., 59, 1917–1942, https://doi.org/10.1175/1520-0469(2002)059<1917:APPFTS>2.0.CO;2, 2002. a
Tost, H., Jöckel, P., Kerkweg, A., Sander, R., and Lelieveld, J.: Technical note: A new comprehensive SCAVenging submodel for global atmospheric chemistry modelling, Atmos. Chem. Phys., 6, 565–574, https://doi.org/10.5194/acp-6-565-2006, 2006a. a, b, c, d, e
Tost, H., Jöckel, P., and Lelieveld, J.: Influence of different convection parameterisations in a GCM, Atmos. Chem. Phys., 6, 5475–5493, https://doi.org/10.5194/acp-6-5475-2006, 2006b. a
Tost, H., Jöckel, P., and Lelieveld, J.: Lightning and convection parameterisations – uncertainties in global modelling, Atmos. Chem. Phys., 7, 4553–4568, https://doi.org/10.5194/acp-7-4553-2007, 2007. a
Tost, H., Lawrence, M. G., Brühl, C., Jöckel, P., The GABRIEL Team, and The SCOUT-O3-DARWIN/ACTIVE Team: Uncertainties in atmospheric chemistry modelling due to convection parameterisations and subsequent scavenging, Atmos. Chem. Phys., 10, 1931–1951, https://doi.org/10.5194/acp-10-1931-2010, 2010. a, b, c
Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., Wagner, R., Simon, M., Lehtipalo, K., Williamson, C., Craven, J. S., Duplissy, J., Adamov, A., Almeida, J., Bernhammer, A.-K., Breitenlechner, M., Brilke, S., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Gysel, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Keskinen, H., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lawler, M., Leiminger, M., Mathot, S., Möhler, O., Nieminen, T., Onnela, A., Petäjä, T., Piel, F. M., Miettinen, P., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Sipilä, M., Smith, J. N., Steiner, G., Tomè, A., Virtanen, A., Wagner, A. C., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Carslaw, K. S., Curtius, J., Dommen, J., Kirkby, J., Kulmala, M., Riipinen, I., Worsnop, D. R., Donahue, N. M., and Baltensperger, U.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 533, 527–531, 2016. a
Ueda, S., Miura, K., Kawata, R., Furutani, H., Uematsu, M., Omori, Y., and Tanimoto, H.: Number–size distribution of aerosol particles and new particle formation events in tropical and subtropical Pacific Oceans, Atmos. Environ., 142, 324–339, https://doi.org/10.1016/j.atmosenv.2016.07.055, 2016. a
Van Dingenen, R., Raes, F., Putaud, J.-P., Baltensperger, U., Charron, A., Facchini, M.-C., Decesari, S., Fuzzi, S., Gehrig, R., Hansson, H.-C., Harrison, R. M., Hüglin, C., Jones, A. M., Laj, P., Lorbeer, G., Maenhaut, W., Palmgren, F., Querol, X., Rodriguez, S., Schneider, J., ten Brink, H., Tunved, P., Tørseth, K., Wehner, B., Weingartner, E., Wiedensohler, A., and Wåhlin, P.: A European aerosol phenomenology-1: physical characteristics of particulate matter at kerbside, urban, rural and background sites in Europe, Atmos. Environ., 38, 2561–2577, https://doi.org/10.1016/j.atmosenv.2004.01.040, 2004. a, b
van Noije, T. P. C., Le Sager, P., Segers, A. J., van Velthoven, P. F. J., Krol, M. C., Hazeleger, W., Williams, A. G., and Chambers, S. D.: Simulation of tropospheric chemistry and aerosols with the climate model EC-Earth, Geosci. Model Dev., 7, 2435–2475, https://doi.org/10.5194/gmd-7-2435-2014, 2014. a, b
Vehkamäki, H., Kulmala, M., Napari, I., Lehtinen, K. E. J., Timmreck, C., Noppel, M., and Laaksonen, A.: An improved parameterization for sulfuric acid-water nucleation rates for tropospheric and stratospheric conditions, J. Geophys. Res.-Atmos., 107, 4622–4631, 2002. a, b
Vehkamäki, H., Kulmala, M., Napari, I., Lehtinen, K. E. J., Timmreck, C., Noppel, M., and Laaksonen, A.: Correction to ”An improved parameterization for sulfuric acid/water nucleation rates for tropospheric and stratospheric conditions”, J. Geophys. Res.-Atmos., 118, 9330–9330, https://doi.org/10.1002/jgrd.50603, 2013. a, b
Wang, H., Easter, R. C., Rasch, P. J., Wang, M., Liu, X., Ghan, S. J., Qian, Y., Yoon, J.-H., Ma, P.-L., and Vinoj, V.: Sensitivity of remote aerosol distributions to representation of cloud-aerosol interactions in a global climate model, Geosci. Model Dev., 6, 765–782, https://doi.org/10.5194/gmd-6-765-2013, 2013. a
Wang, M., Penner, J. E., and Liu, X.: Coupled IMPACT aerosol and NCAR CAM3 model: Evaluation of predicted aerosol number and size distribution, J. Geophys. Res.-Atmos., 114, D06302, https://doi.org/10.1029/2008JD010459, 2009. a, b
Wang, X., Zhang, L., and Moran, M. D.: Uncertainty assessment of current size-resolved parameterizations for below-cloud particle scavenging by rain, Atmos. Chem. Phys., 10, 5685–5705, https://doi.org/10.5194/acp-10-5685-2010, 2010. a
Weinzierl, B., Petzold, A., Esselborn, M., Wirth, M., Rasp, K., Kandler, K., Schütz, L., Koepke, P., and Fiebig, M.: Airborne measurements of dust layer properties, particle size distribution and mixing state of Saharan dust during SAMUM 2006, Tellus B, 61, 96–117, https://doi.org/10.1111/j.1600-0889.2008.00392.x, 2009. a
Weinzierl, B., Ansmann, A., Prospero, J. M., Althausen, D., Benker, N., Chouza, F., Dollner, M., Farrell, D., Fomba, W. K., Freudenthaler, V., Gasteiger, J., Groß, S., Haarig, M., Heinold, B., Kandler, K., Kristensen, T. B., Mayol-Bracero, O. L., Müller, T., Reitebuch, O., Sauer, D., Schäfler, A., Schepanski, K., Spanu, A., Tegen, I., Toledano, C., and Walser, A.: The Saharan Aerosol Long-Range Transport and Aerosol-Cloud-Interaction Experiment: Overview and Selected Highlights, Bull. Am. Meteorol. Soc., 98, 1427–1451, https://doi.org/10.1175/BAMS-D-15-00142.1, 2017. a, b
Winebrake, J. J., Corbett, J. J., Green, E. H., Lauer, A., and Eyring, V.: Mitigating the Health Impacts of Pollution from Oceangoing Shipping: An Assessment of Low-Sulfur Fuel Mandates, Environ. Sci. Technol., 43, 4776–4782, https://doi.org/10.1021/es803224q, 2009. a
Wofsy, S. C., Daube, B. C., Jimenez, R., Kort, E., Pittman, J. V., Park, S., Commane, R., Xiang, B., Santoni, G., Jacob, D., Fisher, J., Pickett-Heaps, C., Wang, H., Wecht, K., Wang, Q.-Q., Stephens, B. B., Shertz, S., Watt, A., Romashkin, P., Campos, T., Haggerty, J., Cooper, W. A., Rogers, D., Beaton, S., Hendershot, R., Elkins, J. W., Fahey, D. W., Gao, R. S., Moore, F., Montzka, S. A., Schwarz, J. P., Perring, A. E., Hurst, D., Miller, B. R., Sweeney, C., Oltmans, S., Nance, D., Hintsa, E., Dutton, G., Watts, L. A., Spackman, J. R., Rosenlof, K. H., Ray, E. A., Hall, B., Zondlo, M. A., Diao, M., Keeling, R., Bent, J., Atlas, E. L., Lueb, R., and Mahoney., M. J.: HIPPO Merged 10-second Meteorology, Atmospheric Chemistry, Aerosol Data (R_20121129), Tech. rep., Carbon Dioxide Information Analysis Center, Oak Ridge National Laboratory, Oak Ridge, Tennessee, USA, https://doi.org/10.3334/CDIAC/hippo_010, 2012. a
Yeatman, S. G., Spokes, L. J., and Jickells, T. D.: Comparisons of coarse-mode aerosol nitrate and ammonium at two polluted coastal sites, Atmos. Environ., 35, 1321–1335, https://doi.org/10.1016/S1352-2310(00)00452-0, 2001. a
Yoon, J., Burrows, J. P., Vountas, M., von Hoyningen-Huene, W., Chang, D. Y., Richter, A., and Hilboll, A.: Changes in atmospheric aerosol loading retrieved from space-based measurements during the past decade, Atmos. Chem. Phys., 14, 6881–6902, https://doi.org/10.5194/acp-14-6881-2014, 2014. a
Young, L.-H., Lee, S.-H., Kanawade, V. P., Hsiao, T.-C., Lee, Y. L., Hwang, B.-F., Liou, Y.-J., Hsu, H.-T., and Tsai, P.-J.: New particle growth and shrinkage observed in subtropical environments, Atmos. Chem. Phys., 13, 547–564, https://doi.org/10.5194/acp-13-547-2013, 2013. a
Zhang, K., O'Donnell, D., Kazil, J., Stier, P., Kinne, S., Lohmann, U., Ferrachat, S., Croft, B., Quaas, J., Wan, H., Rast, S., and Feichter, J.: The global aerosol-climate model ECHAM-HAM, version 2: sensitivity to improvements in process representations, Atmos. Chem. Phys., 12, 8911–8949, https://doi.org/10.5194/acp-12-8911-2012, 2012. a, b
Zhao, C., Chen, S., Leung, L. R., Qian, Y., Kok, J. F., Zaveri, R. A., and Huang, J.: Uncertainty in modeling dust mass balance and radiative forcing from size parameterization, Atmos. Chem. Phys., 13, 10733–10753, https://doi.org/10.5194/acp-13-10733-2013, 2013. a
Zhou, C. and Penner, J. E.: Aircraft soot indirect effect on large-scale cirrus clouds: Is the indirect forcing by aircraft soot positive or negative?, J. Geophys. Res.-Atmos., 119, 11303–11320, https://doi.org/10.1002/2014JD021914, 2014JD021914, 2014. a